Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 1 by Anna Gumieniczek.
Information on absorption, metabolism and excretion of drugs is necessary to support the studies on their pharmacokinetics and potential drug–drug interactions. Moreover, the knowledge on drug metabolism is one of the crucial factors used to assess their pharmacokinetic profile in patients with some dysfunctions. It is especially important in diabetic patients with higher incidence of chronic liver and kidney problems.
- drug metabolism and drug degradation
- chromatographic and radiometric methods
- dedicated packings
- DPP-4
1. Introduction
Gliptins have been developed to prevent degradation of endogenously released incretins, glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide 1 (GLP-1), by DPP-4 enzyme. Thus, they act as DPP-4 inhibitors and prolong respective actions of these endogenous incretins. They do not pass the blood–brain barrier, have no direct effect on satiety, and in contrast with GLP-1 receptor agonists (GLP-1 RAs), did not alter gastric emptying. However, when compared with GLP-1 RAs, the DPP-4 inhibitors offer several advantages such as oral administration, absence of gastrointestinal adverse effects and lower costs. What is more, some clinical data suggest that gliptins could exert positive cardiovascular effects. Because of their safety profile, especially their very low risk of hypoglycemia, gliptins could be indicated in elderly patients [6,7][1][2].
Five of these DPP-4 inhibitors, i.e., anagliptin (ANA, N-[2-[[2-[(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl]amino]-2-methylpropyl]-2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide) (approved in Japan), alogliptin (ALO, 2-{[6-(3-aminopiperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl]methyl}benzonitrile), linagliptin (LINA, 8-[(3R)-3-aminopiperidin-1-yl]-7-but-2-ynyl-3-methyl-1-[(4-methylquinazolin-2-yl)methyl]purine-2,6-dione), sitagliptin (SITA, (3S)-3-amino-1-[3-(trifluoromethyl)-5,6-dihydro[1,2,4][3][4][5]triazolo[4,3-a]pyrazin-7(8H)-yl]-4-(2,4,5-trifluorophenyl)butan-1-one), saxagliptin (SAXA, (1S,3S,5S)-2-[(2S)-2-amino-2-(3-hydroxyadamantan-1-yl)acetyl]-2-azabicyclo[3.1.0]hexane-3-carbonitrile) and vildagliptin (VILDA, (2S)-1-{[(3-hydroxyadamantan-1-yl)amino]acetyl}pyrrolidine-2-carbonitrile) (FDA approved), teneligliptin (TENE, [(2S,4S)-4-[4-(5-methyl-2-phenylpyrazol-3-yl)piperazin-1-yl]pyrrolidin-2-yl]-(1,3-thiazolidin-3-yl)methanone) (approved in Japan, South Korea and India [16) were introduced by regulatory authorities between 2006 and 2013, while evogliptin (EVO, (3R)-4-[(3R)-3-amino-4-(2,4,5-trifluorophenyl)butanoyl]-3-[(2-methylpropan-2-yl)oxymethyl]piperazin-2-one) was approved in South Korea in 2015. Omarigliptin (OMA, (2R,3S,5R)-2-(2,5-difluorophenyl)-5-[2-(methanesulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]oxan-3-amine) and trelagliptin (TRELA, 2-({6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl}methyl)-4-fluorobenzonitrile) are the DPP-4 inhibitors approved in Japan in 2015 as the first once-weekly oral antidiabetic agents in the world [8][6].
Gliptins can be classified into peptidomimetic (i.e., ANA, OMA, SAXA, SITA, TENE and VILDA) and non-peptidomimetic (i.e., ALO, EVO, LINA and TRELA) subtypes. Due to their specificity to the substrate site of the enzyme, some of them have substituted pyrrolidines or thiazolidines as a proline mimetic moiety. Although their chemical structures differ from each other, all DPP-4 inhibitors are substrate-competitive active site binders and have common interactions with the key residues of the target protein [7][2]. Based on the half-life and time of dissociation from the DPP-4 enzyme, they are prescribed twice a day (e.g., ANA and VILDA), once a day (e.g., ALO, EVO, LINA, SITA, SAXA) or once a week (e.g., OMA, TRELA) [6][1]. The structures of the mentioned DPP-4 inhibitors are presented in Figure 1.
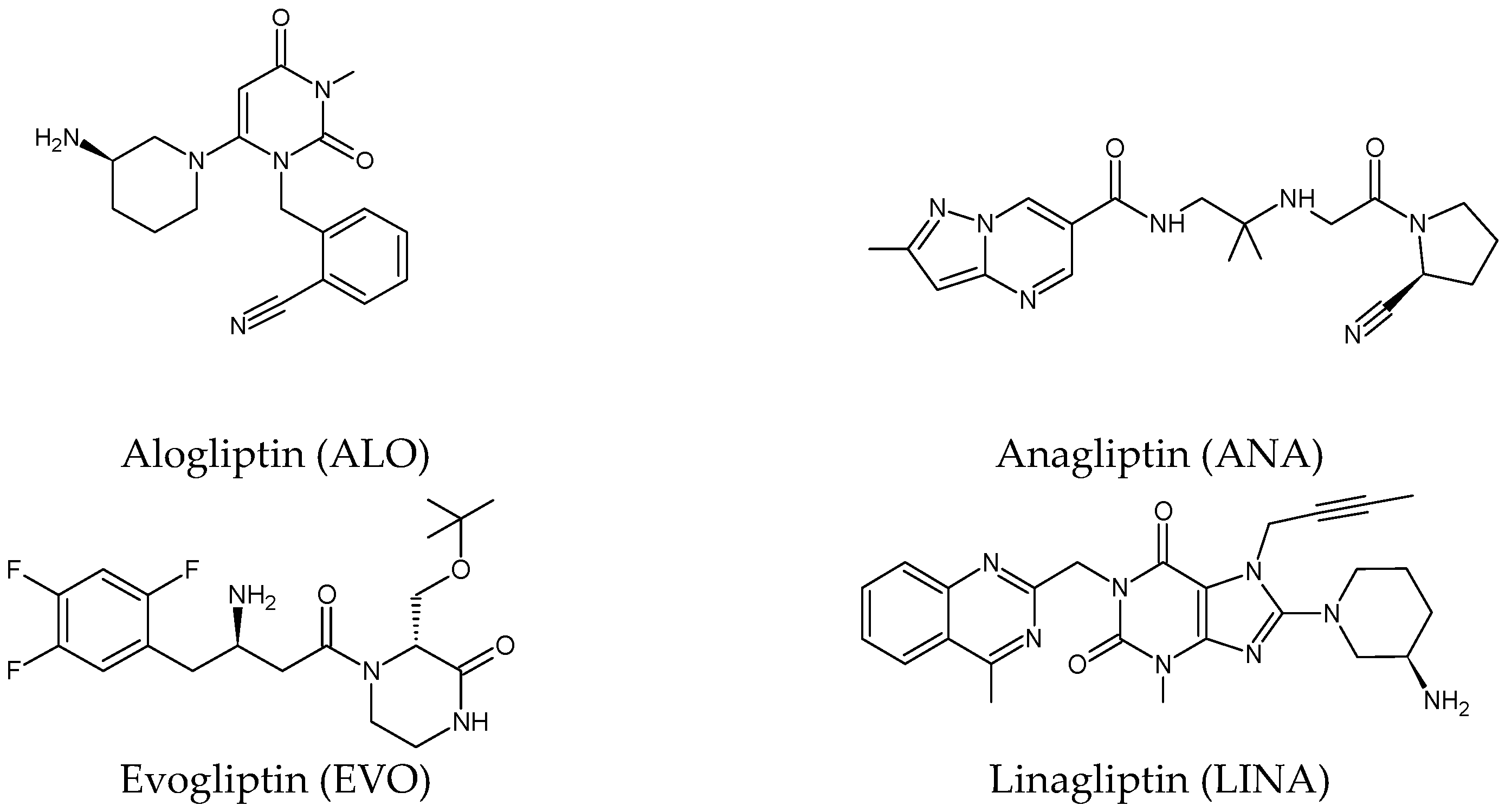
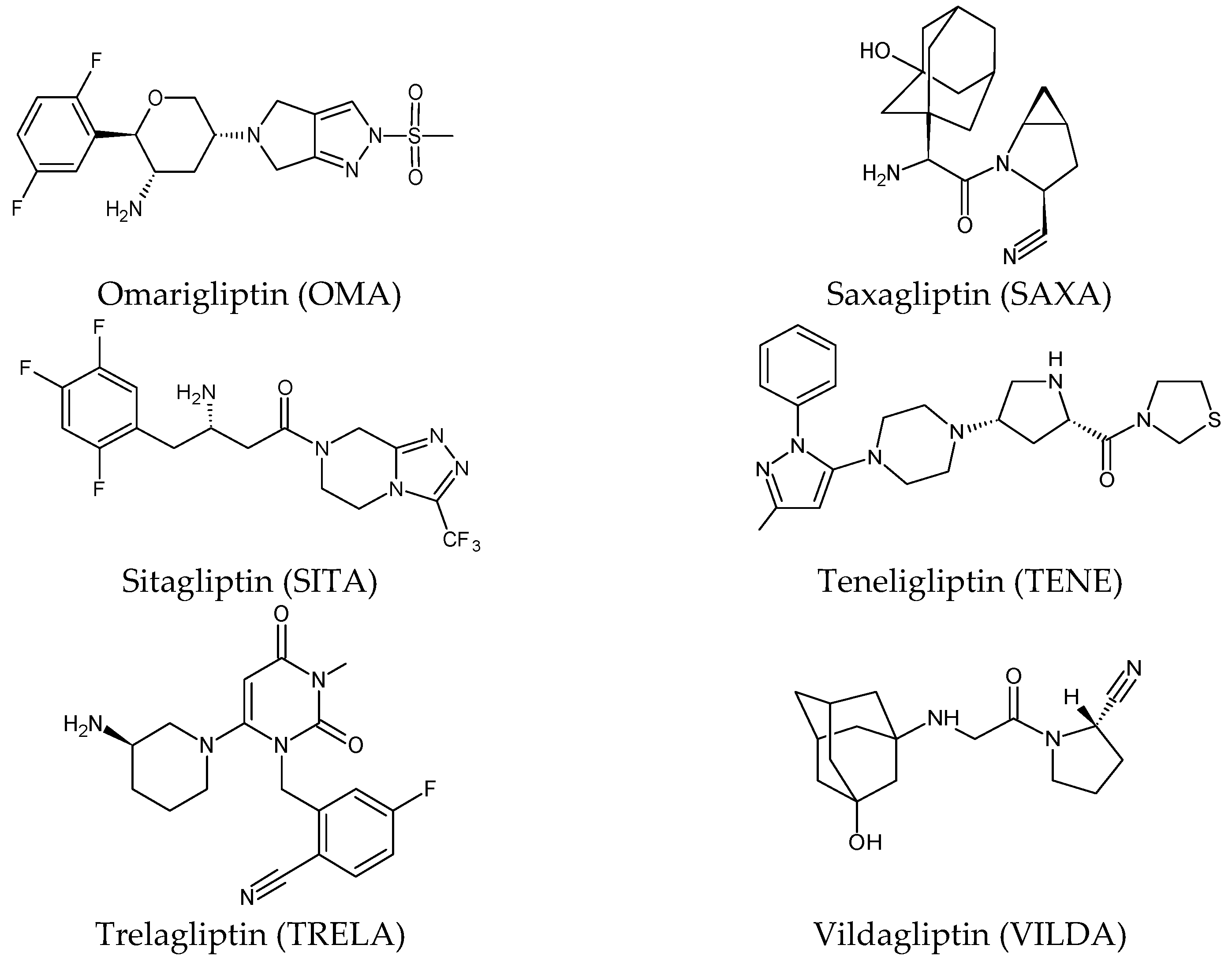
Figure 1. Chemical structures of DPP-4 inhibitors: alogliptin (ALO), anagliptin (ANA), evogliptin (EVO), linagliptin (LINA), omarigliptin (OMA), saxagliptin (SAXA), sitagliptin (SITA), teneligliptin (TENE), trelagliptin (TRELA) and vildagliptin (VILDA).
2. Metabolic Transformations of Gliptins (DPP-4 Inhibitors) and the Methods Used for Elucidating Their Metabolic Pathways
2.1. Metabolism of Gliptins
Alogliptin (ALO) is a highly potent and selective inhibitor of DPP-4 that was developed using the technology of structure-based drug design (SbDD). In the paper from the literature [9][7], it was shown that ca. 10% of ALO is metabolized, while 60–70% of the dose is excreted as unchanged drug in the urine. Two minor metabolites were detected following oral administration of [14C]-ALO, i.e., N-demethylated ALO (ALO-M1) (<1% of the dose) (Table 1) that inhibits DPP-4 similar to the parent molecule, and inactive N-acetylated ALO (<6% of the dose). It was also speculated that ALO is mainly metabolized by CYP2D6 and CYP3A4.
Because anagliptin (ANA) is not frequently used in other countries than Japan, only a small number of reports exists investigating its biological properties and pharmacokinetics. The major metabolic pathway of ANA was proposed as the cyano group hydrolysis to generate the carboxylic acid metabolite ANA-M1 (Table 2), which accounted for 29.2% of the dose. The parent ANA and ANA-M1 were eliminated mainly with urine where the mechanisms of the active transport were probably involved [10][8].
More information on the possible metabolic pathways of evogliptin (EVO) in humans was reported. Metabolism of EVO was proposed as the phase I reactions where the drug is metabolized to 4-oxo-EVO (EVO-M1), 4(S)-hydroxy-EVO (EVO-M2) and 4(R)-hydroxy-EVO (EVO-M3) (Table 1), and as the II phase reactions forming 4(S)-hydroxy-EVO glucuronides and EVO-N-sulfates. At the same time, formation of EVO-M2 and EVO-M3 was inhibited by the CYP3A4 antibody, suggesting that CYP3A4 played a major role in the metabolism of EVO. It was also shown that EVO-M2 could be further metabolized to respective glucuronides by UDP-glucuronosyltransferases UGT2B4 and UGT2B7 [11][9].
The above data on EVO were confirmed in the next study from the literature [12][10]. The metabolism of EVO was also shown by the phase I reactions, i.e., hydroxylation and oxidation, as well as by opening of piperazine ring and oxidation, forming EVO-M4, EVO-M5 and EVO-M6 (Table 1). In addition, the phase II reactions were shown to be involved, i.e., glucuronidation, sulfation, and conjugation with glycine–cysteine moieties.
The chemical structure of linagliptin (LINA) is based on xanthine moiety that distinguishes this drug from other gliptins. It may offer some differences in pharmacokinetic and pharmacodynamic properties of LINA, e.g., its low dissociation from the enzyme and greater potency than other gliptins. It is known that LINA is predominantly eliminated unchanged after both oral and intravenous administration [7][2]. However, the inactive metabolite LINA-M1 was identified in plasma as a major metabolite after oral administration. A two-step mechanism was proposed for its formation, i.e., CYP3A4-dependent conversion of the secondary amine of the parent drug to the corresponding ketone via oxidative deamination, followed by reduction. In excreta, the main metabolite after oral and intravenous administration was LINA-M2 formed by hydroxylation of the methyl group of the butinyl side chain. Next, minor metabolites could be formed by combinations of the following reactions: oxidation of the butinyl side chain and the piperidine moiety followed by oxidative degradation of the piperidine moiety, and finally N-acetylation and glucuronidation. The oxidation of the methyl group at position 4 of the quinazoline moiety was also proposed, and resulted in the corresponding carboxylic acid metabolite LINA-M3 (Table 1). A cysteine adduct and its sulfate conjugate were additionally observed in urine after intravenous administration of LINA [13][11].
In the study of Xu et al. [14][12], absorption, metabolism and excretion of omarigliptin (OMA) were evaluated in healthy male subjects after a single oral dose of 25 mg of [14C]-OMA. Radioactivity levels in plasma and excreta were determined via accelerator mass spectrometry (AMS). As a result, minimal metabolism of OMA was observed, as indicated by the fact that the parent drug accounted for ca. 89% of the radioactivity in urine. However, some oxidative metabolites were detected in plasma, with each comprising less than 3% of the total radioactivity.
Table 1. Metabolites of DPP-4 inhibitors produced by the phase I reactions: alogliptin (ALO), anagliptin (ANA), evogliptin (EVO), linagliptin (LINA), saxagliptin (SAXA), sitagliptin (SITA), teneligliptin (TENE) and vildagliptin (VILDA).
| Metabolite | Structure | m | / | z | [M + H] | + | Ref. |
|---|---|---|---|---|---|---|---|
| ALO-M1 | 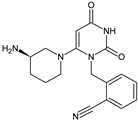 |
n.a. | [9] | [7] | |||
| ANA-M1 | 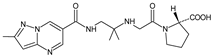 |
366 * | [10] | [8] | |||
| EVO-M1 | 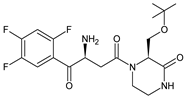 |
416 * | [11] | [9] | |||
| EVO-M2 EVO-M3 (isomers) |
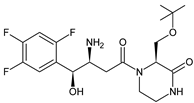 |
418 * | [11,12] | [9][10] | |||
| EVO-M4 | 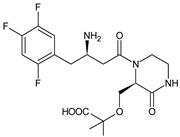 |
418 * | [12] | [10] | |||
| EVO-M5 | 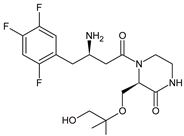 |
432 * | [12] | [10] | |||
| EVO-M6 | 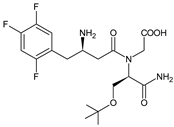 |
434 * | [12] | [10] | |||
| LINA-M1 | 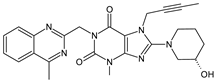 |
n.a. | [7] | [2] | |||
| LINA-M2 | 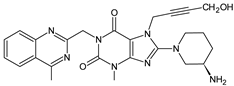 |
n.a. | [13] | [11] | |||
| LINA-M3 | 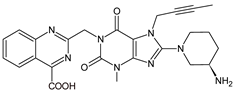 |
n.a. | [13] | [11] | |||
| SAXA-M1 | 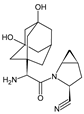 |
332 * | [15,16,17] | [13][14][15] | |||
| SITA-M1 | 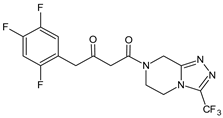 |
408 * | [18] | [16] | |||
| SITA-M2 | 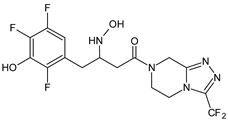 |
422 * | [18] | [16] | |||
| SITA-M3 | 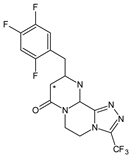 |
406 * | [19] | [17] | |||
| TENE-M1 | 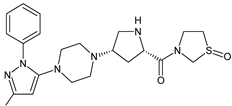 |
443 * | [20,21] | [18][19] | |||
| TENE-M2 | 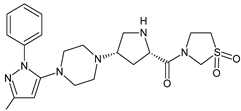 |
n.a. | [20] | [18] | |||
| TENE-M3 | 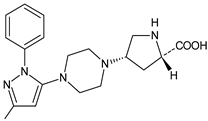 |
n.a. | [20] | [18] | |||
| TENE-M4 | 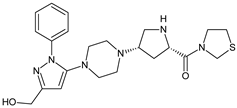 |
n.a. | [20] | [18] | |||
| TENE-M5 | 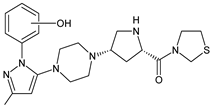 |
n.a. | [20] | [18] | |||
| VILDA-M1 | 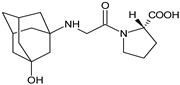 |
n.a. | [7] | [2] |
n.a.—not available in the literature; *—low-resolution LC-MS.
A few metabolites of sitagliptin (SITA) were detected, e.g., diketone metabolite SITA-M1. In addition, hydroxylation of both amine group and aromatic ring followed by formation of glucuronide metabolites, as well as oxidation of NH2 and hydroxylation followed by loss of HF to form SITA-M2, was proposed (Table 1). On the other hand, it was clearly shown that only 3.1% of the parent drug was metabolized over 2 h incubation [19][17].
In the next paper concerning SITA, Vincent et al. [19][17] reported the phase I and phase II reactions of [14C]-SITA and detected its six metabolites in human plasma, urine and feces, using radiometric detection and LC-MS/MS, using the TurboIonSpray® interface operated in positive ion mode. The metabolites were shown to be the products of hydroxylation and N-sulfation of SITA. In addition, oxidation of piperazine ring followed by cyclization was proposed to form SITA-M3 (Table 1). Glucuronidation was also included in SITA metabolism, but it was observed that glucuronides were further transformed and were not detected in feces.
Similar experiments elaborated for teneligliptin (TENE) indicated that 20–34% of the absorbed TENE was excreted through kidneys and 66–80% was metabolized. At the same time, multiple elimination pathways of TENE were proposed, which include metabolic reactions through CYP3A4 and flavin-containing monooxygenase 3 (CYP3A4/FMO3). The identified metabolites were described as TENE-M1 (thiazolidine-1-oxide), TENE-M2 (thiazolidine-1,1-dioxide), TENE-M3 (carboxylic acid formed by hydrolysis of the amide bond), TENE-M4 (hydroxymethyl metabolite formed by oxidation of the methyl group on pyrazole) and TENE-M5 (hydroxylated metabolite with the hydroxyl group on the phenyl moiety) (Table 1) [20][18].
As far as the next drug from gliptins, i.e., vildagliptin (VILDA) is concerned, its metabolism accounts for near 70% of the dose and is the dominant elimination pathway in humans. The major metabolite VILDA-M1 (Table 1) is pharmacologically inactive and is the product of hydrolysis of the cyano moiety. Next, identified metabolites of VILDA were formed by glucuronidation. At the same time, it was shown that the kidneys may be one of the major organs contributing to the hydrolysis of VILDA to VILDA-M1 [7][2].
In the next two studies from the literature [22,23][20][21], it was investigated whether different gliptins, i.e., ANA, ALO, LINA, SITA and VILDA, have the potential to covalently bind to macromolecules and, as a consequence, to initiate immune-mediated hepatotoxicity. As a result, the possibility of covalently binding to macromolecules was shown for ANA and VILDA. It was also found that VILDA and ANA, which both contain a cyanopyrrolidine moiety, rapidly reacted with L-cysteine in no enzymatic manner. Structural analysis of these adducts revealed that the nitrile moieties of both drugs were irreversibly converted to thiazoline acid. Thus, it was found that VILDA and ANA have the potential to covalently bind to a thiol residue of L-cysteine in proteins. Such binding may lead to unpredictable immune responses in humans.
2.2. Analytical Methods for Elucidating the Metabolism of Gliptins
Structural characterization of ANA and its metabolites in humans was performed with radiometric and chromatographic methods (Table 2). Quantitative measurements in respective fractions of mobile phase were taken using a radio flow detector (for urine and feces) or liquid scintillation counter (LSC) (for plasma). Comparing the LC retention times and MS spectra with respective synthetic standards allowed identification of some of the detected metabolites [10][8].
To identify EVO and its metabolites in human liver microsomes, an orbitrap mass spectrometer coupled with the UPLC system was used. Separation was performed on a C8 column using gradient elution. A higher-energy collision dissociation (HCD) system was employed to investigate the fragmentation pattern of EVO and its metabolites (Table 2). As a result, five EVO metabolites were detected and identified via comparison with the retention times and MS/MS spectra of the corresponding standards [11][9]. In addition, a HPLC system coupled with a radioactivity detector (RAD) and an ion trap mass spectrometer (Table 2) was used in order to study of metabolism of [14C]-EVO after oral administration to male rats and dogs [12][10].
In the study of Xu et al. [14][12], absorption, metabolism and excretion of [14C]-OMA were evaluated in healthy male subjects via the AMS method. It is worth noting that AMS shows a special power to separate a rare isotope from an abundant neighboring mass [24][22]. In the mentioned study, the metabolite profiling was conducted using HPLC fractionation followed by AMS to generate radiochromatograms, and finally, metabolites were identified by LC-MS/MS with a hybrid system that consisted of a quadrupole and orthogonal acceleration TOF tandem mass spectrometer (Table 2).
A specific and rapid LC-MS/MS method [15][13] was proposed for simultaneous determination of SAXA and its active metabolite, 5-hydroxy-SAXA (SAXA-M1) in human plasma. Sample preparation was accomplished via solid phase extraction (SPE) using SDS as an ion-pair reagent. In addition, next, the LC-MS/MS method for quantitative determination of SAXA and SAXA-M1 was described in the literature [16][14] (Table 2).
In the study of Peng et al. [17][15], hydrophilic interaction liquid chromatography (HILIC) coupled with mass spectrometry (HILIC-MS) was applied to determine the plasma concentrations of SAXA and SAXA-M1 in the presence of metformin. Chromatographic separation was achieved on a HILIC Chrom Matrix HP amide column that was the optimal choice because of the high differences in hydrophilic properties of the mentioned analytes. Mass spectrometry was performed using a quadrupole tandem mass spectrometer in an MRM mode under positive ESI (Table 2).
A similar analytical strategy was applied in the next study of Khreit et al. [18][16], where the metabolism of SITA was investigated via incubation of the drug with rat hepatocytes. For LC method, a zwitter ionic hydrophilic interaction ZIC-HILIC column with gradient elution and an orbitrap mass spectrometer were used (Table 2).
Additionally, experiments on TENE metabolism were based on the use of AMS technology [20][18]. Metabolite profiling in plasma, urine and feces was performed via offline measurement of radioactivity using a HPLC system. Radioactivity was quantified using a LSC analyzer, with automatic quench correction by an external standard method, while identification of TENE and its metabolites was performed via ESI using a orbitrap hybrid Fourier Transform (FT) mass spectrometer (Table 2). Such hybrid mass spectrometer combines a linear ion trap MS and the orbitrap mass analyzer. Ions generated by a compound are collected and followed by axial ejection to the C-shaped storage trap which is used to store and cool ions before injection into the orbital trap. Signals from each of the orbital trap outer electrodes are amplified and transformed into a frequency spectrum by fast FT, which is finally converted into a mass spectrum [25][23].
Next, a rapid and sensitive LC-MS/MS method for simultaneous quantification of TENE and its active metabolite, TENE-M1, in human plasma was elaborated by Park et al. [21][19], using deuterated TENE as an internal standard. This method was proposed as an alternative to the AMS method described above [20][18], in order to avoid its disadvantages, including its complicated procedure and the expense of designing the radioactive isotope (Table 2).
Table 2. LC and LC-MS methods for determination of metabolites of gliptins: anagliptin (ANA), evogliptin (EVO), linagliptin (LINA), omarigliptin (OMA), saxagliptin (SAXA), sitagliptin (SITA), teneliglitin (TENE) and vildagliptin (VILDA).
| Compound | Conditions | Ref. | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ANA | C18 column (2.0 × 150 mm, 3 μm) and isocratic elution using 1% CH | 3 | COOH/ACN (80:20, | v | / | v | ), 1.0 mL/min; MS/MS with positive ESI. C18 column (4.6 × 250 mm, 5 μm) and gradient elution: (A) 50 mM ammonium acetate, (B) ACN, 1.0 mL/min. |
[10] | [8] | ||||||
| EVO | Unison-C8 column (2.0 × 75 mm, 3.0 μm) and gradient elution of A) 5% ACN in 0.1% HCOOH and B) 95% ACN in 0.1% HCOOH, 0.3 mL/min. The column and autosampler temperatures: 40 °C and 6 °C; ESI in positive and negative mode: spray voltage, 4.0 kV in positive mode and −3.0 kV in negative mode; vaporizer temperature, 350 °C; capillary temperature, 330 °C; sheath gas pressure, 35 Arb; and auxiliary gas pressure, 15 Arb; collision energy of 10 to 40 eV. | [11] | [9] | ||||||||||||
| EVO | Kinetex C18 column (4.6 × 150 mm, 2.6 μm) and gradient elution with (A) 20 mM ammonium acetate (pH 4) and (B) ACN, 1 mL/min. The column temperature 40 °C and UV detection at 268 nm. ESI in the positive mode: spray voltage, 4.5 kV; vaporizer temperature, 350 °C; capillary temperature, 330 °C; sheath gas pressure, 50 Arb; auxiliary gas pressure, 10 Arb; and sweep gas pressure, 5 Arb, the collision gas He, and the normalized collision energy during product ion scanning 35%. |
[12] | [10] | ||||||||||||
| OMA | ACE 5 C8 column (4.6 × 250 mm; 5 µm) and gradient elution with (A) 2 mM ammonium acetate in ACN:H | 2 | O (5:95) containing 0.1% HCOOH, and (B) 2 mM ammonium acetate in ACN:H | 2 | O (95:5) containing 0.1% HCOOH, 1.0 mL/min; N | 2 | as the nebulizer and auxiliary gas, and Ar as the collision gas. The ESI capillary voltage 1.2 kV. The source and desolvation temperatures 100 and 550 °C; collision energy from 20 to 30 eV. |
[14] | [12] | ||||||
| SAXA | ACE CN column (4.6 × 150 mm, 5 μm) and ACN and 10.0 mM ammonium formate buffer of pH 5.0 (80:20, | v | / | v | ); triple quadrupole MS detection with positive ESI. | [15] | [13] | ||||||||
| SAXA | C18 column (2.1 × 50 mm, 5 µm) and gradient elution with (A) 0.1% HCOOH in H | 2 | O and (B) 0.1% HCOOH in ACN. TurboIonSpray | ® | source, positive ionization mode, using SRM. N | 2 | as the nebulizer, curtain and collision gas; 450 °C for TIS interface, 5000 V setting for ion spray voltage, 30 setting for the curtain gas and nebulizer gas. | [16] | [14] | ||||||
| SAXA | HILIC Chrom Matrix HP amide column (3.0 × 100 mm, 5 μm), ACN and 5 mM ammonium formate buffer containing 0.1% HCOOH. Ion spray voltage 5500 V, ion spray temperature 550 °C, ion source gas 1: 50 Arb, ion source gas 2: 55 Arb, curtain gas (N | 2 | ) 30 Arb, collision gas (N | 2 | ) 10 Arb, entrance potential 10 V, collision cell exit potential 12 V. | [17] | [15] | ||||||||
| SITA | ZIC-HILIC column (4.6 mm × 150 mm, 5 µm) and gradient elution with (A) HCOOH in H | 2 | O (0.1% | v | / | v | ) and (B) HCOOH (0.1% | v | / | v | ) in ACN, 0.3 mL/min. The capillary temperature 250 °C, spray voltage +4.5 kV and the sheath and auxiliary gas (N | 2 | ) flow rates 45 and 15. CID voltage of 40 eV. | [18] | [16] |
| TENE | CAPCELL PAK C18 UG120 column (4.6 × 250 mm, 5 µm,) at 40 °C and gradient elution with (A) 20 mM ammonium acetate and (B) ACN, 0.8 mL/min; positive ESI at 3800 V and CID at the collision energy of 35%. | [20] | [18] | ||||||||||||
| TENE | CAPCELL Pak C18 column (2.0 × 2150 mm, 5 μm) and isocratic elution with ACN, MeOH and H | 2 | O, 025 mL/min; temperature of the column and autosampler 50 °C and 10 °C. MS: collision gas 5 psi, curtain gas 10 psi, ion source gas (nebulizer) 30 psi, ion spray voltage 5500 V, and collision energy of 37 eV for TENE; declustering potential, entrance potential, and collision exit potential were 106 V; ESI positive ion mode using MRM. |
[21] | [19] | ||||||||||
| ANA ALO LINA SITA VILDA |
UPLC system and a QQQ mass spectrometer equipped with a switching valve; XBridge C18 column (2.1 × 50 mm, 3.5 μm) and gradient with (A) 0.5 mM ammonium hydrogen carbonate and (B) MeOH for VILDA or (A) 1 mM ammonium acetate and (B) ACN (B) for ANA, ALO, SITA and LINA; 0.55 mL/min. The autosampler 4 °C. ESI positive ion mode using MRM transitions. Orbitrap Fusion MS system coupled with a HPLC system: XBridge C18 column (4.6 × 100 mm, 5 μm) and gradient elution with (A) 20 mM ammonium acetate (A) and (B) ACN (B), 1.0 mL/min; ESI positive and negative ion mode. |
[22,23] | [20][21] |
Arb—arbitrary units.
References
- Rameshrad, M.; Razavi, B.M.; Ferns, G.A.A.; Hosseinzadeh, H. Pharmacology of dipeptidyl peptidase-4 inhibitors and its use in the management of metabolic syndrome: A comprehensive review on drug repositioning. J. Pharm. Sci. 2019, 27, 341–360.
- Chen, X.-W.; He, Z.-X.; Zhou, Z.-W.; Yang, T.; Zhang, X.; Yang, Y.-X.; Duan, W.; Zhou, S.-F. Clinical pharmacology of dipeptidyl peptidase 4 inhibitors indicated for the treatment of type 2 diabetes mellitus. Clin. Exp. Pharmacol. Physiol. 2015, 42, 999–1024.
- Yao, M.; Chen, B.; Zhao, W.; Mehl, J.T.; Li, L.; Zhu, M. LC-MS differential analysis for fast and sensitive determination of biotransformation of therapeutic proteins. Drug Metab. Dispos. 2018, 46, 451–457.
- Rakusanova, S.; Cajka, T. Current analytical methods to monitor type 2 diabetes medication in biological samples. TrAC Trends Anal. Chem. 2023, 158, 116831.
- ICH Topic Q 3 A (R2) “Impurities in New Drug Substances”. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-3-r2-impurities-new-drug-substances-step-5_en.pdf (accessed on 10 May 2023).
- Ahrén, B. DPP-4 Inhibition and the Path to Clinical Proof. Front. Endocrinol. 2019, 10, 376.
- Kaku, K.; Kisanuki, K.; Shibata, M.; Oohira, T. Benefit-risk assessment of alogliptin for the treatment of type 2 diabetes mellitus. Drug Saf. 2019, 42, 1311–1327.
- Furuta, S.; Smart, C.; Hackett, A.; Benning, R.; Warrington, S. Pharmacokinetics and metabolism of anagliptin, a novel dipeptidyl peptidase-4 inhibitor, in humans. Xenobiotica 2013, 43, 432–442.
- Jeong, H.U.; Kim, J.H.; Lee, D.Y.; Shim, H.J.; Lee, H.S.; McPhee, D.J. In vitro metabolic pathways of the new anti-diabetic drug evogliptin in human liver preparations. Molecules 2015, 20, 21802–21815.
- Lee, D.Y.; Kim, J.H.; Shim, H.J.; Jeong, H.U.; Lee, H.S. Absorption, metabolism, and excretion of evogliptin tartrate in male rats and dogs. J. Toxicol. Environ. Health—A: Curr. Issues 2018, 81, 453–464.
- ICH Topic Q 3 A (R2) “Impurities in New Drug Substances Blech, S.; Ludwig-Schwellinger, E.; Gräfe-Mody, E.U.; Withopf, B.; Wagner, K. The metabolism and disposition of the oral dipeptidyl peptidase-4 inhibitor, linagliptin, in humans. Drug Metab. Dispos. 2010, 38, 667–678.
- Xu, S.; Tatosian, D.; Mcintosh, I.; Caceres, M.; Matthews, C.; Samuel, K.; Selverian, D.; Kumar, S.; Kauh, E. Absorption, metabolism and excretion of omarigliptin, a once-weekly DPP-4 inhibitor, in humans. Xenobiotica 2018, 48, 584–591.
- Shah, P.A.; Shah, J.V.; Sanyal, M.; Shrivastav, P.S. LC-MS/MS analysis of metformin, saxagliptin and 5-hydroxy saxagliptin in human plasma and its pharmacokinetic study with a fixed-dose formulation in healthy Indian subjects. Biomed. Chromatogr. 2017, 31, e3809.
- Xu, X.S.; Demers, R.; Gu, H.; Christopher, L.J.; Su, H.; Cojocaru, L.; Boulton, D.W.; Kirby, M.; Stouffer, B.; Humphreys, W.G.; et al. Liquid chromatography and tandem mass spectrometry method for the quantitative determination of saxagliptin and its major pharmacologically active 5-monohydroxy metabolite in human plasma: Method validation and overcoming specific and non-specific binding at low concentrations. J. Chromatogr. B 2012, 889–890, 77–86.
- Peng, Y.; Chang, Q.; Yang, N.; Gu, S.; Zhou, Y.; Yin, L.; Aa, J.; Wang, G.; Sun, J. Quantitative determination of metformin, saxagliptin and 5-hydroxy saxagliptin simultaneously by hydrophilic interaction liquid chromatography-electrospray ionization mass spectrometry and its application to a bioequivalence study with a single-pill combination in human. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018, 1081–1082, 109–117.
- Khreit, O.I.G.; Grant, H.M.; Henderson, C.; Watson, D.G.; Sutcliffec, O.B. Identification of Novel Metabolic Pathways of Sitagliptin (STG) by LC/MS and LC/MS2 after incubations with rat Hepatocytes. J. Drug Metab. Toxicol. 2016, 7, 220.
- Vincent, S.H.; Reed, J.R.; Bergman, A.J.; Elmore, C.S.; Zhu, B.; Xu, S.; Ebel, D.; Larson, P.; Zeng, W.; Chen, L.; et al. Metabolism and excretion of the dipeptidyl peptidase 4 inhibitor sitagliptin in humans. Drug Metab. Dispos. 2007, 35, 533–538.
- Nakamaru, Y.; Hayashi, Y.; Ikegawa, R.; Kinoshita, S.; Madera, B.P.; Gunput, D.; Kawaguchi, A.; Davies, M.; Mair, S.; Yamazaki, H.; et al. Metabolism and disposition of the dipeptidyl peptidase IV inhibitor teneligliptin in humans. Xenobiotica 2014, 44, 242–253.
- Park, J.-W.; Kim, K.-A.; Park, J.-Y. Development of a liquid chromatography/tandem-mass spectrometry assay for the simultaneous determination of teneligliptin and its active metabolite teneligliptin sulfoxide in human plasma. Biomed. Chromatogr. 2020, 34, e4721.
- Mizuno, K.; Takeuchi, K.; Umehara, K.; Nakajima, M. Identification of a novel metabolite of vildagliptin in humans: Cysteine targets the nitrile moiety to form a thiazoline ring. Biochem. Pharmacol. 2018, 156, 312–321.
- Keck, B.D.; Ognibene, T.; Vogel, J.S. Analytical validation of accelerator mass spectrometry for pharmaceutical development. Bioanalysis 2010, 2, 469–485.
- Mizuno, K.; Takeuchi, K.; Umehara, K.; Nakajima, M. Identification of novel metabolites of vildagliptin in rats: Thiazoline-containing thiol adducts formed via cysteine or glutathione conjugation. Drug Metab. Dispos. 2019, 47, 809–817.
- Makarov, A.; Denisov, E.; Kholomeev, A.; Balschun, W.; Lange, O.; Strupat, K.; Horning, S. Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal. Chem. 2006, 78, 2113–2120.
More