Metals are indispensable for the life of all organisms, and their dysregulation leads to various disorders due to the disruption of their homeostasis. Nowadays, various transition metals are used in pharmaceutical products as diagnostic and therapeutic agents because their electronic structure allows them to adjust the properties of molecules differently from organic molecules. Therefore, interest in the study of metal–drug complexes from different aspects has been aroused, and numerous approaches have been developed to characterize, activate, deliver, and clarify molecular mechanisms. The integration of these different approaches, ranging from chemoproteomics to nanoparticle systems and various activation strategies, enables the understanding of the cellular responses to metal drugs, which may form the basis for the development of new drugs and/or the modification of currently used drugs. The purpose of this review is to briefly summarize the recent advances in this field by describing the technological platforms and their potential applications for identifying protein targets for discovering the mechanisms of action of metallodrugs and improving their efficiency during delivery.
- metallodrugs
- medicinal chemistry
- chemoproteomics
- thermal proteome profiling
1. Introduction
2. Protein Target Identification through Proteomic Approaches
The study of protein interactions with different classes of biomolecules has ancient roots in the field of proteomics, with many different biochemical strategies developed and advanced in conjunction with mass spectrometry techniques [39,40][39][40]. In recent years, several proteomic strategies coupled with mass spectrometry (MS) have emerged as a powerful and systematic approach for the large-scale identification of drug–protein interactions and the elucidation of their associated mechanisms. Chemical proteomics, or chemoproteomics, is primarily devoted to the study of protein–small-molecule interactions and is attracting increasing attention in drug target discovery [44,45][44][45]. Chemoproteomics is a leading tool in the field of drug discovery that relies on affinity-based or label-free approaches to identify protein targets.2.1. Affinity-Based Strategies
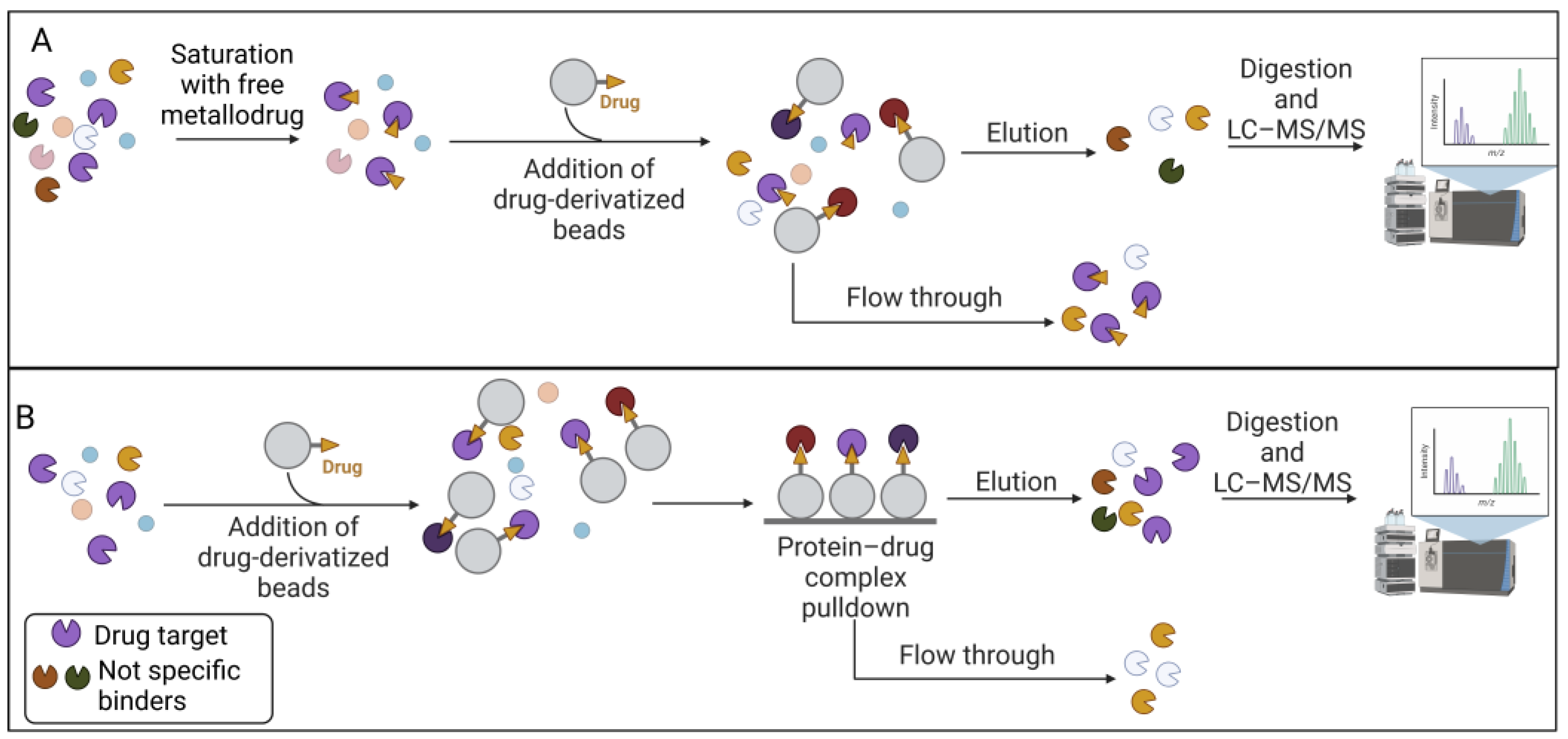
The affinity-based approach to chemical proteomics aims to immobilize the drug on a solid support and isolate the target proteins. This strategy is also referred to as drug pulldown [46,47][46][47]. Pulldown strategies and, more generally, affinity purification-mass spectrometry approaches (AP-MS), are proteomic techniques that allow uresearchers to isolate multiprotein complexes starting from a known molecule to hypothesize the intracellular processes in which it is involved [48,49,50][48][49][50]. Drug pulldown requires the following four main steps: (1) the immobilization of the pharmacophore on resin beads with affinity chromatography; (2) the isolation of the target molecules; (3) the identification of the target molecules with liquid chromatography–tandem mass spectrometry (LC–MS/MS); and (4) bioinformatics analysis to identify and quantify the proteins (Figure 1).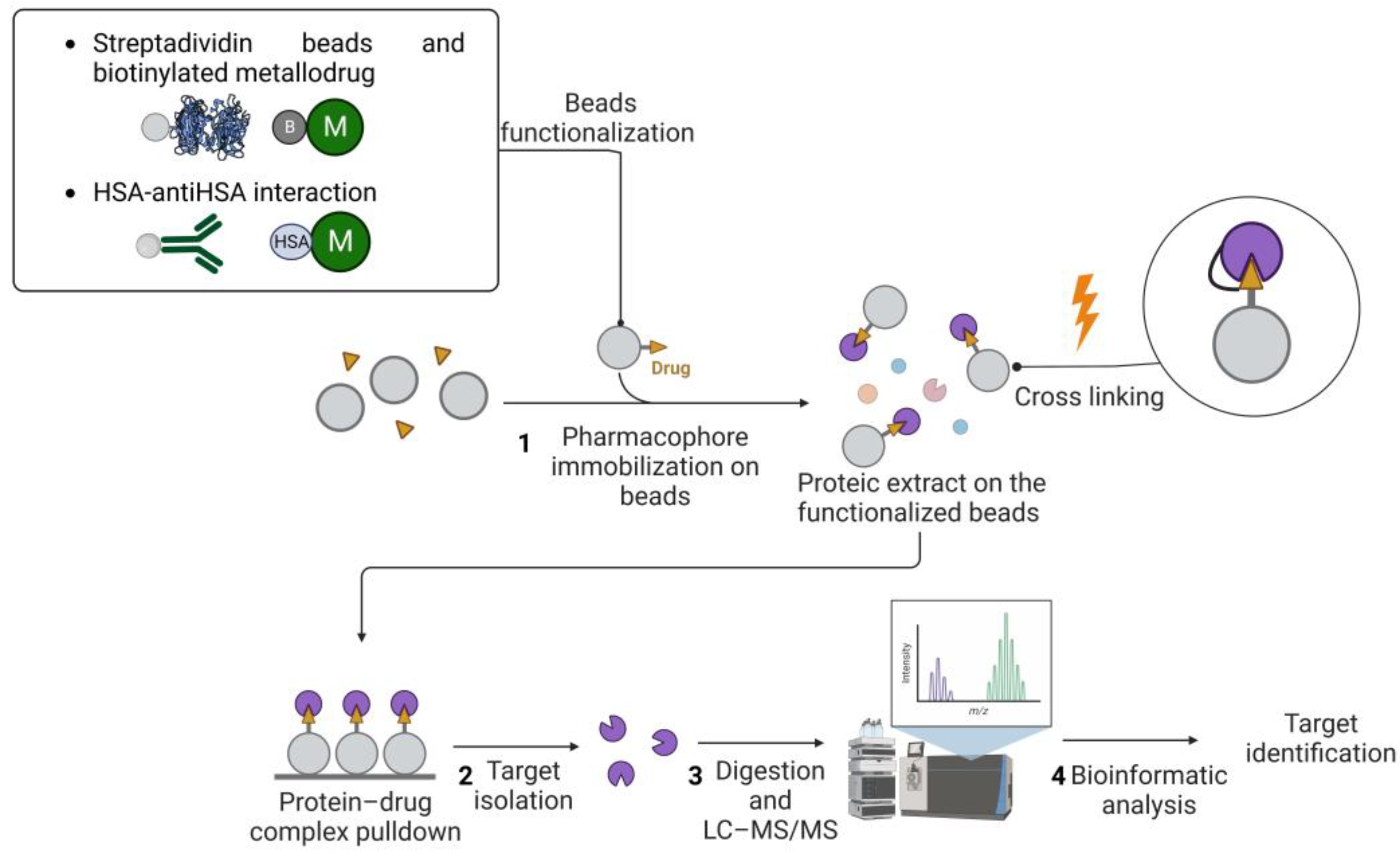
2.2. Label-Free Approaches
On the other hand, label-free approaches exploit protein stability due to the drug interaction. For example, thermal proteome profiling (TPP) [62] has been used to study metallodrug targets [63]. TPP measures the extent of the drug–target interaction by monitoring the effects of pharmacological treatment on protein denaturation/solubility as a function of a progressive increase in temperature. The extent of protein stabilization by the small molecule is proportional to the strength of the interaction and can be assessed by multiplexed mass spectrometry analysis (see [64] for a comprehensive description of all TPP applications). As shown in Figure 3, TPP can be performed by varying the temperature range (TR-TPP) (Figure 3A) or drug concentration (CR-TPP) (Figure 3B). After protein digestion and mass spectrometry analysis using LC–MS/MS, proteins are relatively quantified by the comparison between the drug-treated and drug-free conditions. The soluble fraction for each protein is reported as a function of the temperature or drug concentration.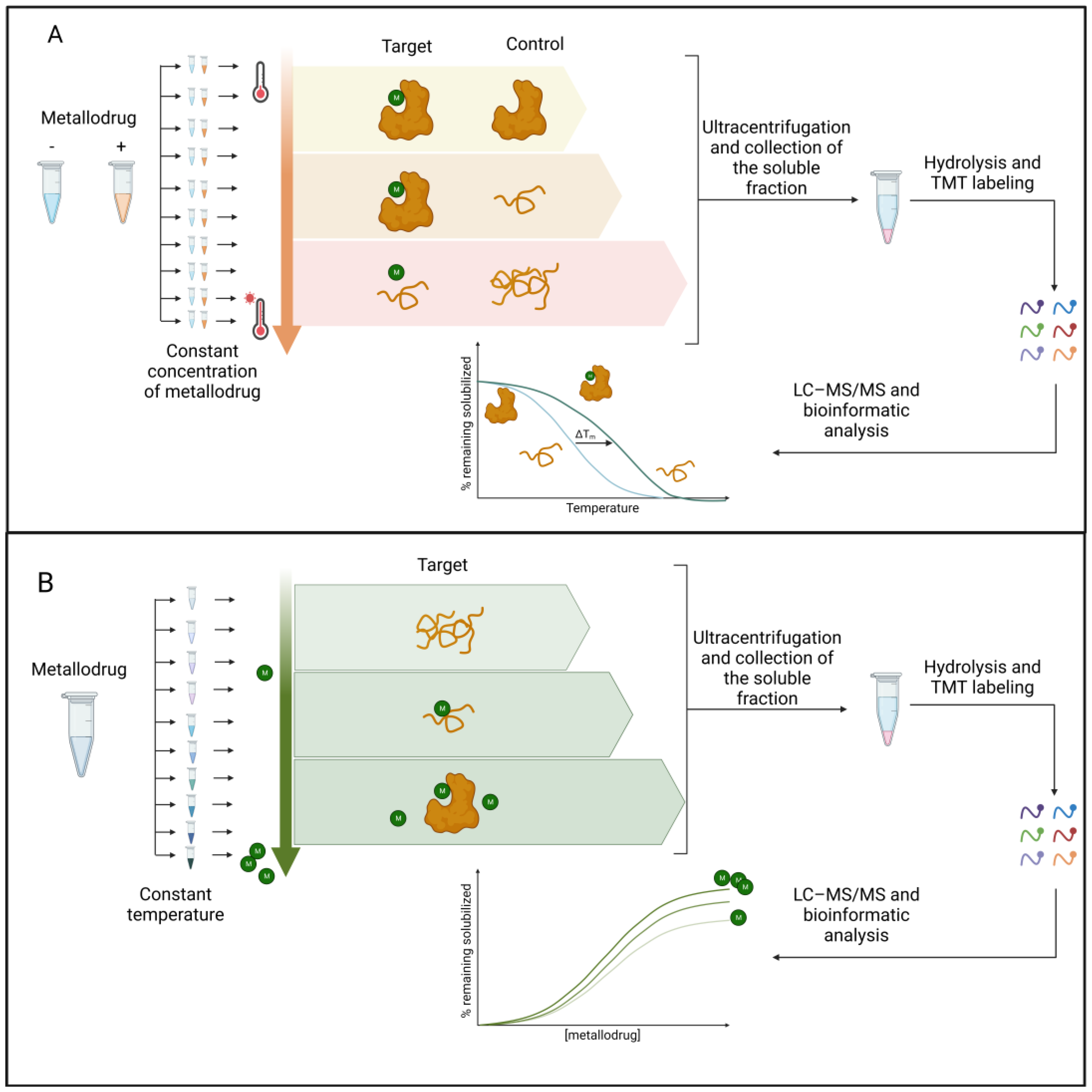
References
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiol. Rev. 2015, 95, 749–784.
- Bagheri-Mohammadi, S.; Farjami, M.; Suha, A.J.; Zarch, S.M.A.; Najafi, S.; Esmaeili, A. The Mitochondrial Calcium Signaling, Regulation, and Cellular Functions: A Novel Target for Therapeutic Medicine in Neurological Disorders. J. Cell. Biochem. 2023, 124, 635–655.
- An, Y.; Li, S.; Huang, X.; Chen, X.; Shan, H.; Zhang, M. The Role of Copper Homeostasis in Brain Disease. Int. J. Mol. Sci. 2022, 23, 13850.
- Ruiz, L.M.; Libedinsky, A.; Elorza, A.A. Role of Copper on Mitochondrial Function and Metabolism. Front. Mol. Biosci. 2021, 8, 711227.
- Opazo, C.M.; Greenough, M.A.; Bush, A.I. Copper: From Neurotransmission to Neuroproteostasis. Front. Aging Neurosci. 2014, 6, 143.
- Wang, L.; Yin, Y.-L.; Liu, X.-Z.; Shen, P.; Zheng, Y.-G.; Lan, X.-R.; Lu, C.-B.; Wang, J.-Z. Current Understanding of Metal Ions in the Pathogenesis of Alzheimer’s Disease. Transl. Neurodegener. 2020, 9, 10.
- Haywood, S. Brain–Barrier Regulation, Metal (Cu, Fe) Dyshomeostasis, and Neurodegenerative Disorders in Man and Animals. Inorganics 2019, 7, 108.
- Franz, K.J.; Metzler-Nolte, N. Introduction: Metals in Medicine. Chem. Rev. 2019, 119, 727–729.
- Galib; Mashru, M.; Patgiri, B.; Barve, M.; Jagtap, C.; Prajapati, P. Therapeutic Potentials of Metals in Ancient India: A Review through Charaka Samhita. J. Ayurveda Integr. Med. 2011, 2, 55.
- Begley, T.P. Wiley Encyclopedia of Chemical Biology; John Wiley & Sons: Hoboken, NJ, USA, 2007; ISBN 978-0-470-04867-2.
- Sodhi, R.K. Metal Complexes in Medicine: An Overview and Update from Drug Design Perspective. Cancer Ther. Oncol. Int. J. 2019, 14, 555883.
- Rosenberg, B.; Van Camp, L.; Krigas, T. Inhibition of Cell Division in Escherichia Coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 205, 698–699.
- Kauffman, G.B.; Pentimalli, R.; Doldi, S.; Hall, M.D. Michele Peyrone (1813–1883), Discoverer of Cisplatin. Platin. Met. Rev. 2010, 54, 250–256.
- Sigel, A.; Sigel, H.; Freisinger, E.; Sigel, R.K.O. (Eds.) Metallo-Drugs: Development and Action of Anticancer Agents; (Metal Ions in Life Sciences); De Gruyter: Berlin, Germany; Boston, MA, USA, 2018; ISBN 978-3-11-046984-4.
- Khoury, A.; Deo, K.M.; Aldrich-Wright, J.R. Recent Advances in Platinum-Based Chemotherapeutics That Exhibit Inhibitory and Targeted Mechanisms of Action. J. Inorg. Biochem. 2020, 207, 111070.
- Zhong, T.; Yu, J.; Pan, Y.; Zhang, N.; Qi, Y.; Huang, Y. Recent Advances of Platinum-Based Anticancer Complexes in Combinational Multimodal Therapy. Adv Healthc. Mater. 2023, 2300253.
- Wang, X.; Wang, X.; Guo, Z. Functionalization of Platinum Complexes for Biomedical Applications. Acc. Chem. Res. 2015, 48, 2622–2631.
- Barabas, K.; Milner, R.; Lurie, D.; Adin, C. Cisplatin: A Review of Toxicities and Therapeutic Applications. Vet. Comp. Oncol. 2008, 6, 1–18.
- Rosenberg, B.; Vancamp, L.; Trosko, J.E.; Mansour, V.H. Platinum Compounds: A New Class of Potent Antitumour Agents. Nature 1969, 222, 385–386.
- Williams, C.J.; Whitehouse, J.M. Cis-Platinum: A New Anticancer Agent. BMJ 1979, 1, 1689–1691.
- Moreno-Alcántar, G.; Picchetti, P.; Casini, A. Gold Complexes in Anticancer Therapy: From New Design Principles to Particle-Based Delivery Systems. Angew. Chem. Int. Ed. 2023, 62, e202218000.
- Coffetti, G.; Moraschi, M.; Facchetti, G.; Rimoldi, I. The Challenging Treatment of Cisplatin-Resistant Tumors: State of the Art and Future Perspectives. Molecules 2023, 28, 3407.
- Florio, D.; La Manna, S.; Annunziata, A.; Iacobucci, I.; Monaco, V.; Di Natale, C.; Mollo, V.; Ruffo, F.; Monti, M.; Marasco, D. Ruthenium Complexes Bearing Glucosyl Ligands Are Able to Inhibit the Amyloid Aggregation of Short Histidine-Peptides. Dalton Trans. 2023, 52, 8549–8557.
- Paul, N.P.; Galván, A.E.; Yoshinaga-Sakurai, K.; Rosen, B.P.; Yoshinaga, M. Arsenic in Medicine: Past, Present and Future. Biometals 2023, 36, 283–301.
- Florio, D.; Iacobucci, I.; Ferraro, G.; Mansour, A.M.; Morelli, G.; Monti, M.; Merlino, A.; Marasco, D. Role of the Metal Center in the Modulation of the Aggregation Process of Amyloid Model Systems by Square Planar Complexes Bearing 2-(2’-Pyridyl)Benzimidazole Ligands. Pharmaceuticals 2019, 12, 154.
- Jaouen, G.; Metzler-Nolte, N.; Alberto, R. (Eds.) Medicinal Organometallic Chemistry; (Topics in Organometallic Chemistry); Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2010; ISBN 978-3-642-13184-4.
- Zhou, Y.; Li, H.; Sun, H. Metalloproteomics for Biomedical Research: Methodology and Applications. Annu. Rev. Biochem. 2022, 91, 449–473.
- Kostova, I. Ruthenium Complexes as Anticancer Agents. Curr. Med. Chem. 2006, 13, 1085–1107.
- Huang, H.; Cao, K.; Kong, Y.; Yuan, S.; Liu, H.; Wang, Y.; Liu, Y. A Dual Functional Ruthenium Arene Complex Induces Differentiation and Apoptosis of Acute Promyelocytic Leukemia Cells. Chem. Sci. 2019, 10, 9721–9728.
- Ahrweiler-Sawaryn, M.-C.; Biswas, A.; Frias, C.; Frias, J.; Wilke, N.L.; Wilke, N.; Berkessel, A.; Prokop, A. Novel Gold(I) Complexes Induce Apoptosis in Leukemia Cells via the ROS-Induced Mitochondrial Pathway with an Upregulation of Harakiri and Overcome Multi Drug Resistances in Leukemia and Lymphoma Cells and Sensitize Drug Resistant Tumor Cells to Apoptosis in Vitro. Biomed. Pharmacother. 2023, 161, 114507.
- Ferraro, M.G.; Piccolo, M.; Misso, G.; Santamaria, R.; Irace, C. Bioactivity and Development of Small Non-Platinum Metal-Based Chemotherapeutics. Pharmaceutics 2022, 14, 954.
- Peña, Q.; Wang, A.; Zaremba, O.; Shi, Y.; Scheeren, H.W.; Metselaar, J.M.; Kiessling, F.; Pallares, R.M.; Wuttke, S.; Lammers, T. Metallodrugs in Cancer Nanomedicine. Chem. Soc. Rev. 2022, 51, 2544–2582.
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The Side Effects of Platinum-Based Chemotherapy Drugs: A Review for Chemists. Dalton Trans. 2018, 47, 6645–6653.
- Casini, A.; Vessieres, A.; Meier-Menches, S.M. (Eds.) Metal-Based Anticancer Agents; (Metallobiology Series); Royal Society of Chemistry: London, UK, 2019; ISBN 978-1-78801-406-9.
- Mjos, K.D.; Orvig, C. Metallodrugs in Medicinal Inorganic Chemistry. Chem. Rev. 2014, 114, 4540–4563.
- Barhamand, B.A. Difficulties Encountered in Implementing Guidelines for Handling Antineoplastics in the Physician’s Office. Cancer Nurs. 1986, 9, 138–143.
- Zanca, C.; Cozzolino, F.; Quintavalle, C.; Di Costanzo, S.; Ricci-Vitiani, L.; Santoriello, M.; Monti, M.; Pucci, P.; Condorelli, G. PED Interacts with Rac1 and Regulates Cell Migration/Invasion Processes in Human Non-Small Cell Lung Cancer Cells. J. Cell. Physiol. 2010, 225, 63–72.
- Fusco, S.; Aulitto, M.; Iacobucci, I.; Crocamo, G.; Pucci, P.; Bartolucci, S.; Monti, M.; Contursi, P. The Interaction between the F55 Virus-Encoded Transcription Regulator and the RadA Host Recombinase Reveals a Common Strategy in Archaea and Bacteria to Sense the UV-Induced Damage to the Host DNA. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2020, 1863, 194493.
- Iacobucci, I.; Monaco, V.; Cozzolino, F.; Monti, M. From Classical to New Generation Approaches: An Excursus of -Omics Methods for Investigation of Protein-Protein Interaction Networks. J. Proteom. 2021, 230, 103990.
- Cozzolino, F.; Iacobucci, I.; Monaco, V.; Monti, M. Protein–DNA/RNA Interactions: An Overview of Investigation Methods in the -Omics Era. J. Proteome Res. 2021, 20, 3018–3030.
- Kumara, B.N.; Kalimuthu, P.; Prasad, K.S. Synthesis, Properties and Potential Applications of Photoluminescent Carbon Nanoparticles: A Review. Anal. Chim. Acta 2023, 1268, 341430.
- Xia, Y.; Fu, S.; Ma, Q.; Liu, Y.; Zhang, N. Application of Nano-Delivery Systems in Lymph Nodes for Tumor Immunotherapy. Nano-Micro Lett. 2023, 15, 145.
- Kargozar, S.; Moghanian, A.; Rashvand, A.; Miri, A.K.; Hamzehlou, S.; Baino, F.; Mozafari, M.; Wang, A.Z. Nanostructured Bioactive Glasses: A Bird’s Eye View on Cancer Therapy. WIREs Nanomed. Nanobiotechnol. 2023, e1905.
- Fedorov, I.I.; Lineva, V.I.; Tarasova, I.A.; Gorshkov, M.V. Mass Spectrometry-Based Chemical Proteomics for Drug Target Discoveries. Biochemistry 2022, 87, 983–994.
- Skos, L.; Borutzki, Y.; Gerner, C.; Meier-Menches, S.M. Methods to Identify Protein Targets of Metal-Based Drugs. Curr. Opin. Chem. Biol. 2023, 73, 102257.
- Steel, T.R.; Hartinger, C.G. Metalloproteomics for Molecular Target Identification of Protein-Binding Anticancer Metallodrugs. Metallomics 2020, 12, 1627–1636.
- Ziegler, S.; Pries, V.; Hedberg, C.; Waldmann, H. Target Identification for Small Bioactive Molecules: Finding the Needle in the Haystack. Angew. Chem. Int. Ed. 2013, 52, 2744–2792.
- Iacobucci, I.; Monaco, V.; Canè, L.; Bibbò, F.; Cioffi, V.; Cozzolino, F.; Guarino, A.; Zollo, M.; Monti, M. Spike S1 Domain Interactome in Non-Pulmonary Systems: A Role beyond the Receptor Recognition. Front. Mol. Biosci. 2022, 9, 975570.
- Federico, A.; Sepe, R.; Cozzolino, F.; Piccolo, C.; Iannone, C.; Iacobucci, I.; Pucci, P.; Monti, M.; Fusco, A. The Complex CBX7-PRMT1 Has a Critical Role in Regulating E-Cadherin Gene Expression and Cell Migration. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2019, 1862, 509–521.
- Cozzolino, F.; Vezzoli, E.; Cheroni, C.; Besusso, D.; Conforti, P.; Valenza, M.; Iacobucci, I.; Monaco, V.; Birolini, G.; Bombaci, M.; et al. ADAM10 Hyperactivation Acts on Piccolo to Deplete Synaptic Vesicle Stores in Huntington’s Disease. Hum. Mol. Genet. 2021, 30, 1175–1187.
- Babak, M.V.; Meier, S.M.; Huber, K.V.M.; Reynisson, J.; Legin, A.A.; Jakupec, M.A.; Roller, A.; Stukalov, A.; Gridling, M.; Bennett, K.L.; et al. Target Profiling of an Antimetastatic RAPTA Agent by Chemical Proteomics: Relevance to the Mode of Action. Chem. Sci. 2015, 6, 2449–2456.
- Wang, X.; Zhu, M.; Gao, F.; Wei, W.; Qian, Y.; Liu, H.-K.; Zhao, J. Imaging of a Clickable Anticancer Iridium Catalyst. J. Inorg. Biochem. 2018, 180, 179–185.
- Wang, X.; Zhang, J.; Zhao, X.; Wei, W.; Zhao, J. Imaging and Proteomic Study of a Clickable Iridium Complex. Metallomics 2019, 11, 1344–1352.
- Neuditschko, B.; King, A.P.; Huang, Z.; Janker, L.; Bileck, A.; Borutzki, Y.; Marker, S.C.; Gerner, C.; Wilson, J.J.; Meier-Menches, S.M. An Anticancer Rhenium Tricarbonyl Targets Fe−S Cluster Biogenesis in Ovarian Cancer Cells. Angew. Chem. Int. Ed. 2022, 61, e202209136.
- Neuditschko, B.; Legin, A.A.; Baier, D.; Schintlmeister, A.; Reipert, S.; Wagner, M.; Keppler, B.K.; Berger, W.; Meier-Menches, S.M.; Gerner, C. Interaction with Ribosomal Proteins Accompanies Stress Induction of the Anticancer Metallodrug BOLD-100/KP1339 in the Endoplasmic Reticulum. Angew. Chem. Int. Ed. 2021, 60, 5063–5068.
- Li, L.; Zhang, Z. Development and Applications of the Copper-Catalyzed Azide-Alkyne Cycloaddition (CuAAC) as a Bioorthogonal Reaction. Molecules 2016, 21, 1393.
- Wang, Y.; Li, H.; Sun, H. Metalloproteomics for Unveiling the Mechanism of Action of Metallodrugs. Inorg. Chem. 2019, 58, 13673–13685.
- Hu, D.; Liu, Y.; Lai, Y.-T.; Tong, K.-C.; Fung, Y.-M.; Lok, C.-N.; Che, C.-M. Anticancer Gold(III) Porphyrins Target Mitochondrial Chaperone Hsp60. Angew. Chem. Int. Ed. 2016, 55, 1387–1391.
- Wang, S.; Tian, Y.; Wang, M.; Wang, M.; Sun, G.; Sun, X. Advanced Activity-Based Protein Profiling Application Strategies for Drug Development. Front. Pharmacol. 2018, 9, 353.
- Lu, K. Chemoproteomics: Towards Global Drug Target Profiling. ChemBioChem 2020, 21, 3189–3191.
- Plowright, A.T. Target Discovery and Validation: Methods and Strategies for Drug Discovery; (Methods and Principles in Medicinal Chemistry); Wiley-VCH: Weinheim, Germany, 2020; ISBN 978-3-527-34529-8.
- Savitski, M.M.; Reinhard, F.B.M.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Molina, D.M.; Jafari, R.; Dovega, R.B.; Klaeger, S.; et al. Tracking Cancer Drugs in Living Cells by Thermal Profiling of the Proteome. Science 2014, 346, 1255784.
- Saei, A.A.; Gullberg, H.; Sabatier, P.; Beusch, C.M.; Johansson, K.; Lundgren, B.; Arvidsson, P.I.; Arnér, E.S.J.; Zubarev, R.A. Comprehensive Chemical Proteomics for Target Deconvolution of the Redox Active Drug Auranofin. Redox Biol. 2020, 32, 101491.
- Mateus, A.; Kurzawa, N.; Becher, I.; Sridharan, S.; Helm, D.; Stein, F.; Typas, A.; Savitski, M.M. Thermal Proteome Profiling for Interrogating Protein Interactions. Mol. Syst. Biol. 2020, 16, e9232.
- Hu, D.; Yang, C.; Lok, C.; Xing, F.; Lee, P.; Fung, Y.M.E.; Jiang, H.; Che, C. An Antitumor Bis(N-Heterocyclic Carbene)Platinum(II) Complex That Engages Asparagine Synthetase as an Anticancer Target. Angew. Chem. Int. Ed. 2019, 58, 10914–10918.
- Chernobrovkin, A.; Marin-Vicente, C.; Visa, N.; Zubarev, R.A. Functional Identification of Target by Expression Proteomics (FITExP) Reveals Protein Targets and Highlights Mechanisms of Action of Small Molecule Drugs. Sci. Rep. 2015, 5, 11176.
- Lee, R.F.S.; Chernobrovkin, A.; Rutishauser, D.; Allardyce, C.S.; Hacker, D.; Johnsson, K.; Zubarev, R.A.; Dyson, P.J. Expression Proteomics Study to Determine Metallodrug Targets and Optimal Drug Combinations. Sci. Rep. 2017, 7, 1590.
- Strickland, E.C.; Geer, M.A.; Tran, D.T.; Adhikari, J.; West, G.M.; DeArmond, P.D.; Xu, Y.; Fitzgerald, M.C. Thermodynamic Analysis of Protein-Ligand Binding Interactions in Complex Biological Mixtures Using the Stability of Proteins from Rates of Oxidation. Nat. Protoc. 2013, 8, 148–161.
- Park, C.; Marqusee, S. Pulse Proteolysis: A Simple Method for Quantitative Determination of Protein Stability and Ligand Binding. Nat. Methods 2005, 2, 207–212.
- Lomenick, B.; Jung, G.; Wohlschlegel, J.A.; Huang, J. Target Identification Using Drug Affinity Responsive Target Stability (DARTS). Curr. Protoc. Chem. Biol. 2011, 3, 163–180.
- Feng, F.; Zhang, W.; Chai, Y.; Guo, D.; Chen, X. Label-Free Target Protein Characterization for Small Molecule Drugs: Recent Advances in Methods and Applications. J. Pharm. Biomed. Anal. 2023, 223, 115107.
- Jia, S.; Wang, R.; Wu, K.; Jiang, H.; Du, Z. Elucidation of the Mechanism of Action for Metal Based Anticancer Drugs by Mass Spectrometry-Based Quantitative Proteomics. Molecules 2019, 24, 581.
- Roberts, E.A.; Sarkar, B. Metalloproteomics: Focus on Metabolic Issues Relating to Metals. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 425–430.