Under the umbrella of cutaneous sarcomas (CS), a heterogeneous group of rare, malignant, mesenchymal neoplasia, such as dermatofibrosarcoma protuberans, atypical fibroxanthoma, cutaneous undifferentiated pleomorphic sarcoma, cutaneous angiosarcoma and leiomyosarcoma is included. Clinical presentation and histopathological examination are the cornerstone of CS diagnosis and classification. CS are characterized by mostly unspecific dermatoscopic patterns. Dermatofibrosarcoma protuberans, Kaposi’s sarcoma, and in a lesser degree, cutaneous angiosarcoma, may display distinct dermatoscopic features, facilitating their early clinical recognition. In conclusion, dermatoscopy, in conjunction with the overall clinical context, may aid towards suspicion of CS.
1. Introduction
Sarcomas are a wide group of solid, malignant neoplasms of mesenchymal origin, including more than seventy histological variants. They are characterized by significant heterogeneity in regards to the age of onset, localization, biologic behavior, disease course and prognosis. The vast majority of sarcomas emerge from the soft tissues, whilst about 20% arise in bones. Based on their incidence that ranges between 4 and 5 per 100,000 per year in Europe, they are considered a rare type of cancer
[1][2][3][4][1,2,3,4].
Under the term cutaneous sarcomas (CS), a plethora of malignant mesenchymal neoplasia that originate either in the dermis or subcutis is included
[1]. The most common entities comprise dermatofibrosarcoma protuberans (DFSP), atypical fibroxanthoma (AFX) and cutaneous undifferentiated pleomorphic sarcoma (CUPS). Kaposi’s sarcoma (KS) and angiosarcoma represent the two major sarcomas of vascular origin involving the dermis and subcutis. Other less common neoplasms are cutaneous liposarcoma and leiomyosarcoma. The majority of CS are characterized by a comparatively good prognosis, as they mostly tend to recur locally, with low rates of distant metastases
[1][2][3][4][1,2,3,4].
CS are far less common compared to other skin malignancies, such as keratinocyte carcinomas and cutaneous melanoma. Delayed diagnosis, with subsequent delays in therapeutic interventions, is a very common scenario in CS
[3]. The latter is not only related to their rarity, but can be also attributed to their highly atypical and variable clinical presentation, the lack of public awareness and the poor experience of healthcare professionals with this type of malignancy.
Dermatoscopy (synonyms: dermoscopy, epiluminescence microscopy, incident light microscopy, skin-surface microscopy) is a non-invasive, in vivo diagnostic technique, initially used for the examination of suspicious cutaneous lesions. Dermatoscopy involves the use of a handheld tool, named dermatoscope. The dermatoscopic examination facilitates visualization of anatomic structures that are invisible by naked eye clinical examination. Anatomic alterations visible under the dermatoscope can be located in the epidermis, the dermo-epidermal junction or the papillary dermis.
23. Dermatofibrosarcoma Protuberans (DFSP)
DFSP is considered the most common type of CS. Its rough estimated incidence ranges from 0.8 to 5 cases per million inhabitants, per year. It affects both males and females, with a peak incidence between the second and fifth decade of life. Sites of predilection are the trunk and extremities, but it may ostensibly involve any area of skin
[5][6][7][8][9][6,7,8,9,10].
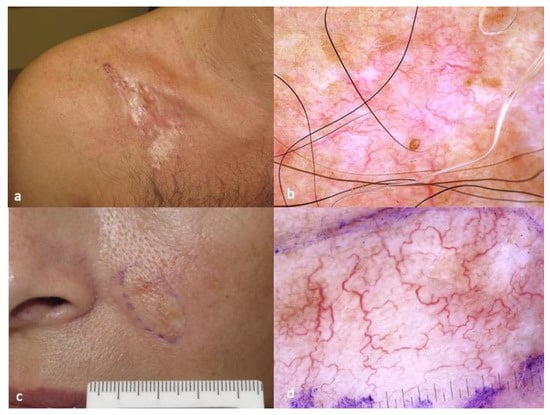
DFSP clinically presents as a firm, exophytic plaque with coalescent, brown-red or flesh-colored nodules on its surface. At the initial stage of evolution, DFSP may present as an atrophic, flat plaque, mimicking a scar or scleroderma. Indolent slow growth, often extending for many years is the rule. During childhood, the predominant clinical morphology is that of a flat or slightly elevated lesion, closely mimicking the plaque-type scleroderma. The size of a DFSP lesion is highly variable, depending on time from disease onset, and usually ranges from 2 to 5 cm in largest diameter. Gigantic lesions measuring up to 20 cm have also been reported. Due to the aforementioned characteristics, delayed diagnosis of DFSP is not uncommon
[5][6][7][8][9][6,7,8,9,10].
The dermoscopic pattern of DFSP has been described in case series and case reports, and a fully developed plaque most often consists of a pink-colored background, structureless depigmented areas, structureless light brown areas and vessels that are mostly linear and arborizing (
Figure 1)
[10][11][12][13][14][15][16][17][18][19][11,12,13,14,15,16,17,18,19,20]. Shiny white streaks and fine pigment network can be also present. The latter findings are not pathognomonic of DFSP and in certain cases may closely mimic a morpheic BCC (
Figure 1)
[10][11][12][13][14][15][16][17][18][19][11,12,13,14,15,16,17,18,19,20].
Figure 1. Typical clinical (a) and dermatoscopic (b) image of DFSP displaying pink-colored background, structureless depigmented areas, structureless light brown areas and linear, arborizing vessels. Morpheic BCC (c) closely mimicking DFSP in dermatoscopy (d). The photos are from the databases of two of the authors (Z.A., A.L.) and all individuals have provided written informed consent for the use of their photos for scientific purposes.
In terms of histopathology, the tumor is characterized by poorly delimited architecture and presence of spindle cells with a storiform to whorled pattern, largely extending into the subcutaneous tissues. There are monomorphous, minimally pleomorphic, spindle cells with long nuclei and low mitotic activity, intercellular collagen, and small capillaries. The irregular cord-like projections of tumor cells, extending far beyond the center of the tumor, are possibly responsible for the high recurrence rates, even after adequate surgical resection.
DFSP has a favorable prognosis, with a reported 10-year survival rate reaching 99%. Treatment of choice is Mohs, with complete micrographic control of excisional margins. If Mohs surgery is not available, surgical excision with wide margins is strongly recommended due to high rates of local recurrence. In the scenario of recurrent fibrosarcomatous DFSP, a CT scan of the lungs is recommended, since the lungs represent the most common site of metastasis.
34. Atypical Fibroxanthoma (AFX)
AFX primarily affects elderly individuals and it is mostly seen in chronically sun-exposed areas of the skin, such as the head/neck area. Prevalence and incidence rates of AFX are vastly unknown due to its rarity.
The precise pathogenesis of AFX is still obscure. Mutations in the p53 tumor suppressor gene typically related to a UV signature are often identified
[1][2][20][21][1,2,21,22].
On the clinical basis, AFX presents as a rapidly enlarging, firm, dome-shaped, red or flesh-colored nodule on sun-exposed areas of the skin in elderly individuals. The surface of the lesion is often eroded or crusted. Differential diagnosis includes benign and malignant skin neoplasms, such as NMSCs, DFSP, amelanotic melanoma and pleomorphic dermal sarcoma (PDS)
[1][2][20][21][22][23][24][25][26][27][1,2,21,22,23,24,25,26,27,28].
While some investigators believe there is no distinction between AFX and PDS, others support that they should be classified as different entities. Analytically, AFX is located in the dermis and superficial subcutis, lacking perineural or vascular invasion, as opposed to PDS, which is infiltrating the deeper layers of subcutis, occasionally displaying perineural and/or vascular infiltration. Currently, AFX is considered a neoplasia of intermediate malignancy, based on its limited metastatic potential
[20][21][22][23][24][25][26][27][21,22,23,24,25,26,27,28].
The dermatoscopic characteristics of AFX are markedly unspecific, simulating other malignant tumors, such as amelanotic melanoma and undifferentiated SCC. As shown in a multicentric study conducted by the International Dermoscopy Society, the majority of AFXs display red and white structureless areas and irregular linear vessels (
Figure 2).
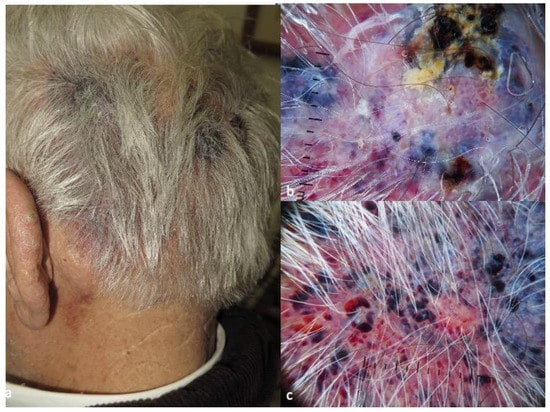
Figure 2. Clinical aspect of patch-type Kaposi’s sarcoma (
a), displaying purple-pink color in the background, white lines, white clods and rosettes in dermatoscopy (
b). Clinical aspect of nodular Kaposi’s sarcoma lesion (
c), characterized by the presence of collaret and polychromatic (“rainbow”) pattern in dermatoscopy (
d). The photos are from the databases of two of the authors (Z.A., A.L.) and all individuals have provided written informed consent for the use of their photos for scientific purposes.
45. Cutaneous Undifferentiated Pleomorphic Sarcoma (CUPS)
CUPS is a rare soft tissue sarcoma with vague etiopathogenesis. Until recently, the tumor was described in the literature as “malignant fibrous histiocytoma”, a term that was abandoned in the last decades
[20][24][25][26][21,25,26,27]. CUPS refers to those CS that cannot be unequivocally classified, despite extensive histological studies. Whether AFX represents the superficial variant of CUPS is still under debate, since both, the histological features and the immunohistochemical profile, literally correspond to those observed in AFX
[1][2][19][27][28][1,2,20,28,29]. The reason of diagnostic distinction between AFX and CUPS is primarily their different biologic behavior, suggesting a higher likelihood of local recurrence, distant metastasis and tumor-related mortality in CUPS as compared to AFX
[27][28].
The Caucasian population is more commonly affected and the age of onset ranges between 50 and 70 years. About 70% of cases occur among men. In terms of topography, the most affected areas are the thighs and the trunk, whilst head and neck are unusual sites, occurring almost exclusively in adults. CUPS clinically manifests as a gradually enlarging subcutaneous nodule that may ultimately acquire a significant size and ulcerate
[1][2][24][25][26][27][28][1,2,25,26,27,28,29].
There are scarce data in the literature concerning the dermatoscopic features of CUPS
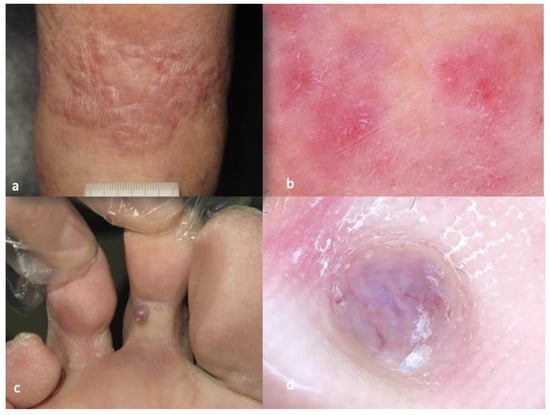
[24][25][26][27][28][25,26,27,28,29]. In the few published cases, it was shown that red and white structureless zones were the predominant features, in combination with thick, linear irregular vessels (
Figure 3).
Figure 3. Clinical aspect of AFX (
a), characterized by red and white structureless areas and irregular linear vessels in dermatoscopy (
b). Clinical aspect of CUPS (
c), showing red and white structureless zones in combination with thick, linear irregular vessels in dermatoscopy (
d). The photos are from the databases of two of the authors (Z.A., A.L.) and all individuals have provided written informed consent for the use of their photos for scientific purposes.
The gold standard of diagnosis is histological and immunohistochemistry studies that facilitate correct diagnosis and discrimination from other entities with similar morphological alterations
[1][2][26][27][1,2,27,28].
56. Kaposi’s Sarcoma (KS)
KS was described in the literature for the first time by a Viennese dermatologist named Moritz Kaposi (1837–1902), about one century ago. Since then, KS has gained much attention among the scientific community, especially after the recognition of its etiologic relation with the acquired immune deficiency syndrome (AIDS). KS is an enigmatic angioproliferative disorder classified into four epidemiological groups, namely AIDS-related (epidemic), iatrogenic (transplant-related), endemic and classic KS. In terms of pathogenesis, all of the aforementioned variants have been linked to the oncogenic virus HHV8 (human herpes virus 8). Classic (or Mediterranean) KS was first described by Kaposi and typically affects middle-aged or elderly individuals. Classic KS has a predilection for males of Mediterranean, East European, or Jewish origin. Endemic KS was originally observed among children and adults in sub-Saharan African populations prior to the arrival of the acquired immunodeficiency syndrome (AIDS).
The clinical presentation of KS is highly heterogenous, with some individuals presenting with latent, indolent disease and others with more aggressive tumors. The skin is the most common site of involvement, followed by mucocutaneous areas, lymph nodes and visceral organs; in rare scenarios, urinary and musculoskeletal systems, heart and eyes, but also endocrine organs may be involved. In regards to the clinical morphology of cutaneous lesions, patch, plaque and nodular type are the most common ones.
The dermatoscopic pattern of KS is greatly related to the clinical morphology of the lesions
[29][30][31][32][33][34][35,36,37,38,39,40]. Analytically, in patches, scholars mostly observe purple-pink color in the background and white lines, white clods and rosettes (
Figure 4). Vessels, present in less than 20% of the cases, are usually dotted and serpentine. Dermatoscopy of plaques is more diverse in terms of vessel morphology and color, with the presence of pink, purple, blue and white. In addition to the white clods and lines, white scales appear in 3 out of 10 KS plaques. Nodular lesions are characterized by the presence of a collaret and polychromatic (“rainbow”) pattern, in about 40% and 50% of cases, correspondingly (
Figure 4).
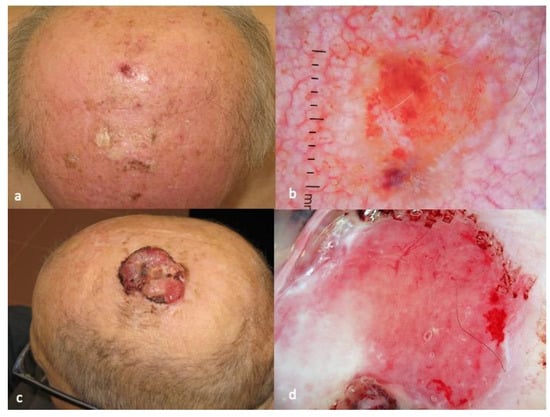
Figure 4. Clinical aspect of CAS (
a), with dark red, blue and purple dots and clods, divided by white lines and focal hemorrhagic clods in dermatoscopy (
b). Focally, the dark red clods may mimic the lacunae of hemangioma (
c). The photos are from the databases of two of the authors (Z.A., A.L.) and all individuals have provided written informed consent for the use of their photos for scientific purposes.
67. Angiosarcoma
Angiosarcoma (AS) is a rare and highly aggressive tumor, comprising less than 2% of all soft tissue sarcomas
[1][2][34][35][36][37][38][39][40][41][42][1,2,40,41,42,43,44,45,46,47,48]. AS originates from lymphatic or vascular endothelial cells and can occur at any age; however, it mainly affects elderly patients. AS comprises a clinically and genetically heterogeneous subgroup of sarcomas and it can occur in any location of the body, with the liver, breast and retroperitoneum being the most common visceral organs affected.
CAS represents less than 5% of all skin cancers. It is considered an orphan disease with an annual incidence of about 0.5–1 cases per million population. The etiology of CAS is not well understood; however, some risk factors have been identified. These include exposure to certain chemicals such as arsenic and vinyl chloride, history of radiation therapy and chronic lymphedema.
CAS usually presents as a rapidly growing, violaceous or reddish nodule or plaque on the skin, most commonly on the scalp, face, ears and neck. It can also occur on the trunk and extremities. The lesion may have a raised, ulcerated or crusted surface. CAS, at its early presentation, can also mimic benign vascular lesions such as hemangioma or pyogenic granuloma
[34][35][36][37][38][39][40][41][42][40,41,42,43,44,45,46,47,48].
The diagnosis of CAS is typically made through a combination of clinical examination, dermatoscopy and histopathologic examination. On dermatoscopy, CAS usually presents with dark red and purple or violaceous structureless zones on a light red background. Dark red, blue and purple dots and clods, divided by thick, perpendicular, white lines, reminding the lacuna of hemangiomas, can also be present. Hemorrhagic clods and white and yellow circles, respectively corresponding to focal ulcerations and follicular plugs/opening.
78. Cutaneous Leiomyosarcoma
Cutaneous leiomyosarcoma (CLMS, synonymous with atypical intradermal smooth muscle neoplasms) is an aggressive, rare type of skin cancer (accounts for less than 1% of all skin cancers) that arises from smooth muscle cells. The majority of CLMS cases occur in individuals over the age of 50 years, with a male-to-female ratio of 2:1. In some cases, a genetic predisposition has been identified in families with multiple cases of CLMS (e.g., TP53 germline mutation carriers)
[1][2][43][44][45][1,2,49,50,51].
CLMS typically presents as a firm, skin-colored, dermal nodule that may be seen anywhere on the body, but most commonly on the extremities, head/neck region and the trunk. It is typically well-circumscribed and can range in size from a few millimeters to several centimeters. The nodules may be fixed to underlying structures and may occasionally be tender to pressure due to their size and the compression of the surrounding tissues. The overlying skin may be normal or slightly erythematous, with occasional ulceration or crusting
[1][2][43][44][45][1,2,49,50,51].
The diagnosis of CLMS is challenging, as it can mimic a plurality of other skin lesions, including epidermal cysts and skin metastases. The first step in the diagnostic process is a thorough clinical examination, including a detailed history and physical examination, with histopathology being the golden standard for diagnosis.
Dermatoscopy has been shown to be a useful tool in the evaluation of CLMS
[46][52]. The dermatoscopic features of CLMS consist of linear or circular structures with a white to yellow color and homogenous or speckled pigmentation. In addition, dermatoscopy may reveal the presence of telangiectasias and brown to reddish dots or globules
[46][52].
The gold standard in diagnosis remains histopathology and immunohistochemistry studies. CLMS histologically displays high cellularity, with presence of atypical spindle cells arranged in fascicles, bundles, or nodules. Positivity in smooth muscle actin, desmin and h-caldesmon may facilitate discrimination of CLMS from other spindle cell tumors. Description of the depth of infiltration is of paramount importance, considering that dermal localization is linked to a poorer prognosis
[1][2][43][44][45][1,2,49,50,51].
89. Cutaneous Liposarcoma
Cutaneous liposarcoma originates from transformed adipocytes and it is considered an extremely rare malignancy. It mostly affects individuals between 50 and 70 years of age and is clinically characterized by the presence of a subcutaneous nodule, closely resembling an epidermal cyst or a banal lipoma. Histopathology is the only way to establish a definite diagnosis. Upon histopathology, cutaneous liposarcoma can be classified as well-differentiated, dedifferentiated, myxoid, and round cell type. Combinations of the aforementioned subtypes can also be seen
[2][47][2,54].
As for the majority of cutaneous sarcomas, wide local excision and histolological examination of the margins is the treatment of choice. Adjuvant radiotherapy can be offered in deep infiltrating tumors, whilst chemotherapy is preserved for advanced, metastatic disease
[2][47][2,54].