Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 4 by Francesco Buonfiglio.
Glaucoma is the most prevalent optic nerve disease worldwide. Oxidative stress plays a pivotal role in the pathophysiology of optic nerve diseases such as glaucoma, Leber’s hereditary optic neuropathy (LHON), and anterior ischemic optic neuropathy (AION)AION. Imbalances between reactive oxygen species (ROS) as well as reactive nitrogen species (RNS) generation and antioxidant systems lead to reactive species overproduction, adenosine triphosphate (ATP) insufficiency, irreversible cellular injuries, and ultimately retinal ganglion cell (RGC)RGC loss.
- oxidative stress
- optic nerve
- retinal ganglion cell
- glaucoma
1. Introduction
Optic nerve diseases encompass a wide range of disorders characterized by optic nerve atrophy, resulting from the loss of retinal ganglion cells (RGCs) and leading to sight-threatening conditions [1][2][3]. These pathologies include:
-
Anterior ischemic optic neuropathies (AION): This category includes arteritic forms, like giant cell arteritis (GCA), which has a pooled prevalence of approximately 51.74 in 100,000 for individuals over the age of 50 [15]. Nonarteritic forms have a reported prevalence of approximately 102.87 in 100,000 in the general population over the age of 40 in the Republic of Korea [16];
-
Infiltrative optic neuropathies, such as leukemic optic neuropathy, which presents in approximately 16% and 18% of all chronic and acute leukemia cases, respectively [28];
Glaucoma is the most prevalent optic nerve disease worldwide [6][7]. LHON has a low estimated prevalence for complete penetrance cases [8][9][10][11][12], while the prevalence is much higher for carriers of mutation variants in the general population [13][14]. The common underlying feature in all optic nerve diseases is the damage and loss of RGCs and their axons, which gradually leads to optic nerve degeneration [3][33][34]. RGCs have high energy requirements and are particularly susceptible to alterations in their energy supply, mainly generated in the mitochondria through the electron transport chain (ETC) [35]. Oxidative stress plays a pivotal role in the pathophysiology of optic nerve diseases such as glaucoma, LHON, and AION. Imbalances between reactive oxygen species (ROS) as well as reactive nitrogen species (RNS) generation and antioxidant systems lead to reactive species overproduction, adenosine triphosphate (ATP) insufficiency, irreversible cellular injuries, and ultimately RGC loss [3][36][37][38][39][40][41][42][43][44][45].
2. Anatomy and Perfusion of the Visual Pathway
The optic nerve, also known as the second cranial nerve [46], is composed of thin (0.1 µm) and lengthy (~50 mm) RGC axons that extend from the retina to the lateral geniculate nucleus, resulting in a soma/axon ratio of approximatively 1:10,000 [35]. Within the retinal layers, these axons merge to form the retinal nerve fiber layer [47], which runs parallel to the superficial blood vessels. The inner retina, including the outer plexiform layer through the nerve fiber layer, is supplied by the central retinal artery. On the other hand, the avascular outer retina, consisting of the outer nuclear layer and photoreceptors, receives its blood supply through diffusion from the choriocapillaris. The choriocapillaris is nourished by short posterior ciliary arteries, which branch from the ophthalmic artery [48][49]. Notably, retinal oxygenation exhibits variability depending on light or dark conditions due to the different oxygen demand of rods and cones. In humans, rods, which are responsible for vision in low light, are present in approximately 120 million, while cones number around 6 million [50]. Consequently, retinal oxygen consumption is reduced by half in the presence of light, attributed to the decreased activity of rods compared to cones [50].
Optic nerve axons account for approximately 38% of all axons within the central nervous system [51]. Around 1.2 million RGC axons converge to form the optic nerve head (ONH), also referred to as the optic papilla or optic disc. The ONH exhibits a brighter central depression known as the optic cup [51][52][53][54]. Blood supply to the ONH in humans is provided by the arterial circle of Zinn–Haller [48].
The optic nerve can be divided into four compartments: the intraocular segment (1–2 mm), which includes the retinal nerve fiber layer (RNFL) and extends from the ONH to the lamina cribrosa; the intraorbital segment (25–30 mm), which spans from the retrobulbar tract to the optic canal; the intracanalicular segment (5–9 mm); and the intracranial segment (9–10 mm), which extends from the optic canal to the optic chiasm [51]. Four distinct regions can be identified within the optic nerve head: the nerve fiber layer, the prelaminar region, the lamina cribrosa, and the retrolaminar region [47]. The lamina cribrosa serves as a supportive structure for the RGC axons within the ONH [55][56] and consists of approximately 200–300 porous apertures through which the optic nerve passes from the sclera into the retrobulbar cavity [51]. Deformations of the lamina cribrosa may indicate RGC loss and can signify the initial stages of glaucomatous optic neuropathy [57]. Once beyond the lamina cribrosa, the optic nerve becomes myelinated by oligodendrocytes, increasing its diameter from 1–2 mm to 3–4 mm [51]. The intraorbital, intracanalicular, and intracranial segments receive their blood supply from the posterior ciliary arteries as well as the circle of Willis [58].
The two optic nerves converge at the optic chiasm, where nerve fibers originating from the nasal retina of each eye cross over to join the temporal fibers of the contralateral eye [47][59][60]. The blood supply to the chiasm is provided by the circle of Willis [47][52]. From the chiasm, the RGC axons continue their course into the optic tract, which receives perfusion from the posterior communicating and internal carotid artery [52]. Within the optic tract, the nerve fibers undergo rearrangement to align with their corresponding positions in the lateral geniculate nucleus [59]. Fibers carrying visual information from the right visual field project to the left cerebral hemisphere and vice versa [59]. In the lateral geniculate nucleus, the RGC axons synapse with the second-order neurons of the visual pathway, organized in six layers consisting mainly of large and small neurons [47]. Some fibers from the optic tract also synapse with the olivary pretectal nucleus, regulating the pupillary light reflex [52][61]. Additionally, RGC axons containing melanopsin terminate in the suprachiasmatic nucleus, a crucial center for controlling the circadian rhythms [47][61].
The large and small axons of the lateral geniculate nucleus form optic radiation, which initially projects anteriorly and then turns posteriorly, terminating in the occipital lobe where the visual cortex (Brodmann area 17) is located [47][59]. These regions receive blood supply from branches of the internal carotid artery (lateral geniculate nucleus and optic radiation) and the posterior cerebral artery (visual cortex) [52]. In Figure 1, the perfusion and the anatomy of the visual pathway are illustrated.
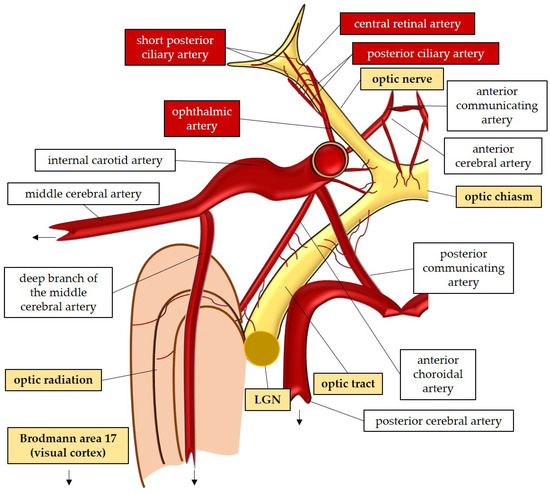
Figure 1. Anatomy and perfusion of the visual pathway. LGN: lateral geniculate nucleus.
An interesting aspect of the optic nerve anatomy is the presence of an unmyelinated portion in the RGCs [35]. In the unmyelinated compartment, specifically in the intraocular segment of the optic nerve, potential signals cannot be transmitted through saltatory conduction due to the absence of myelin [62]. To compensate for this limitation and enable rapid transmission, RGCs generate higher quantities of ATP in their axons to repolarize the plasma membrane [63]. Mitochondrial bidirectional transport (antero- and retrograde) along the axons plays a crucial role in this process. These organelles move toward regions with high energy demands, such as the unmyelinated portions, and ATP gradients are believed to guide this transport [35]. This mechanism may explain the specific vulnerability of RGCs to mitochondrial dysfunction, leading to the triggering of ROS production in a vicious cycle [44][63].
It is important to highlight the trophic role of myelin in the optic nerve sheath. Myelin has been shown to play a vital role in supplying nutrients to the axon, as the entire mitochondrial respiratory chain has been detected in the myelin sheath of the optic nerve [64]. This finding provides possible explanations for the link between myelin loss and axonal degeneration observed in neuropathies or demyelinating disorders [64]. A decrease in myelin-related mitochondrial respiration may be one of the main triggers responsible for neurodegenerative events [65]. With aging, there is a loss of structural integrity in the myelin sheath, which can subsequently lead to axonal deterioration [66]. This process may underlie various neurodegenerative pathologies, including Alzheimer’s disease, as recent studies have suggested [67]. Therefore, an intriguing future therapeutic approach for neurodegenerative disorders involves improving the integrity of the myelin sheath, which could potentially slow disease progression [67]. In this regard, the comanipulation of microglia and a specific signaling pathway, such as the G protein-coupled receptor 17 pathway in oligodendrocyte precursor cells, has been shown to induce robust myelination and promote axonal regeneration following injury [68]. Exploring these avenues may also offer new therapeutic perspectives for other neurodegenerative diseases, including optic neuropathies [69].
3. General Mechanisms of Nitro-Oxidative Stress in the Optic Nerve
3.1. Generation of Reactive Oxygen and Nitrogen Species
Mitochondria are vital intracellular organelles responsible for essential chemical reactions that produce energy substrates [70][71]. In addition to their various cellular functions, such as modulating intracellular calcium levels, synthesizing nucleotides, lipids, and amino acids, and regulating apoptosis, mitochondria also generate ROS [70][72][73][74]. ROS, at basal levels, serve as critical mediators of signaling pathways, including hypoxic and inflammatory pathways [44][70][75][76]. The fundamental function of mitochondria is to regulate oxygen metabolism and produce energy in the form of ATP [70][71][77]. The electron transport chain (ETC) within the inner mitochondrial membrane plays a central role in this process [78]. Despite the efficiency of oxidative phosphorylation, electron leaks can occur, leading to the direct interaction of electron carriers with molecular oxygen (O2) in the mitochondrial matrix. This interaction results in the donation of electrons and the generation of superoxide (O2•−) [71][77][79][80]. While mitochondria are recognized as the main source of ROS in the cell, other significant sources include the enzymatic activities of nitric oxide synthase (NOS) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) [3][79][81][82]. NOS generates nitric oxide (NO), while NOX (comprising seven isoforms: NOX1, -2, -3, -4, -5, DUOX1, -2) transfers electrons from cytosolic NADPH to molecular O2, generating O2•− [83][84]. NO is a free radical that plays a pivotal role in various physiological functions [85]. It serves as a regulator of vascular tone [86][87][88]. Additionally, NO acts as a signaling molecule in neurotransmission and as a regulator of gene transcription [89][90][91][92][93][94]. The production of NO is facilitated by the activity of NOS, an enzyme that has three isoforms: neuronal NOS (nNOS or NOS I); inducible NOS (iNOS or NOS II); and endothelial NOS (eNOS or NOS III) [85][95]. NO rapidly and spontaneously reacts with O2•− through a “diffusion-limited reaction” [96][97]. As a result, a highly damaging RNS termed peroxynitrite (ONOO−) is generated [85][96][97]. Peroxynitrite contributes to the pathogenesis of diverse retinal disorders, being also newly proposed as a critical factor in the pathogenesis of glaucoma [98][99][100].3.2. Oxidative Damage and Antioxidant Defense Systems
ROS and RNS play a physiological role in cellular responses to hypoxia, cell proliferation, cell death, inflammation, or infection [44][76]. Immune cells, such as phagocytes, produce ROS, which provide reactions necessary for an appropriate killing of pathogens [101][102]. Due to endogenous or exogenous trigger factors, the balance between pro- and antioxidant systems can be critically undermined, resulting in nitro-oxidative stress. In this context, radicals begin to compete for paired electrons with intracellular substrates [103], creating oxidative damage. Oxidative injuries are recognized to be a crucial player in the pathogenesis of a variety of pathologies, including ocular diseases [36][104][105][106][107]. At the biomolecular level, three general forms of injuries caused by reactive species can be distinguished: DNA lesions [108][109], protein alterations [110][111], and lipid peroxidation [96][112]. The consequences of DNA damage are modifications in the expression of proteins and the altered regulation of fundamental activities, like oxidative phosphorylation, according to the vicious cycle theory [113][114][115]. In this context, mitochondrial ROS also induce activation of the nod-like receptor family pyrin domain-containing 3 (Nlrp3) inflammasome, a key factor in pyroptotic cell death during inflammation [116]. Antioxidant systems are responsible for defending cells and tissues from the damaging impact of reactive species, which are constantly produced as a “by-product” of oxidative phosphorylation but also serve, at basal levels, physiological functions [76]. Enzymatic antioxidants comprise SOD, catalase (CAT), glutathione peroxidase (GPX), glutathione-S-transferase (GST), heme oxygenase (HO), peroxiredoxin, and thioredoxin [103][117][118][119][120][121][122]. Nonenzymatic antioxidants can be classified into direct and indirect agents. Direct antioxidants react with ROS or RNS, “being sacrificed in the process of their antioxidant actions” [123][124]. Free radical scavengers are, for example, glutathione (GSH) [125], carotenoids [126], vitamin C (ascorbic acid) [127], and vitamin E (α-tocopherol) [128]. Alternatively, indirect antioxidants are molecules, such as vitamin C, that upregulate antioxidant proteins, for example, via the nuclear factor erythroid-2-related factor 2 (Nrf2) [123][127] or molecules, like α-lipoic acid [129]. Examples of antioxidant compounds adsorbed with the food are resveratrol and betulinic acid [98][130][131].3.3. Oxidative Stress in Retinal Ganglion Cells
The retina belongs to the metabolically most active organs in the human body [132] and requires a relatively large amount of energy substrates [133], which makes it particularly vulnerable to energy insufficiency [134]. Oxygen supply is essential for retinal function [135], and its consumption occurs very rapidly, like in the brain [136][137][138]. Hence, conditions that can modify the supply of molecules, such as O2, necessary for the production of energy substrates, like ATP, may rapidly generate significant damage in RGCs due to their susceptibility to oxygen deficiency (Figure 2). Thus, an appropriate blood supply via retrobulbar and retinal vessels is crucial for the proper function of RGCs. Studies in retrobulbar blood vessels reported that ROS blunted endothelial function partially by reducing the contribution of the NOS pathway to endothelium-dependent vasodilation [139]. Likewise, moderately elevated IOP induced endothelial dysfunction in retinal arterioles together with RGC loss [140][141]. Zadeh et al. found in apolipoprotein E (ApoE)-deficient mice that hypercholesterolemia caused oxidative stress and endothelial dysfunction in retinal arterioles but did neither lead to increased ROS levels in the RGC layer nor to loss of RGCs, indicative of compensatory effects [142]. In contrast, a study in pigs reported that after only 12 min of ocular ischemia and 20 h of reperfusion, endothelial dysfunction, retinal edema, and RGC loss occurred [143]. ROS generation due to ischemia/reperfusion (I/R) injury is reported to be caused by diverse enzymes involved in the regulation of oxidative metabolism, such as NOX2, xanthine oxidase (XO), uncoupled eNOS, and by ETC dysfunction [143][144][145][146]. Hyperglycemia was also described to be a cause of endothelial dysfunction and oxidative stress in the retina [147][148][149] via the involvement of NOX2 due to the activation of the receptor of an advanced glycation end product (RAGE)-, mitogen-activated protein kinase (MAPK)-, polyol-, protein kinase C (PKC)-, renin–angiotensin system (RAS) signaling pathways [150][151][152][153][154][155].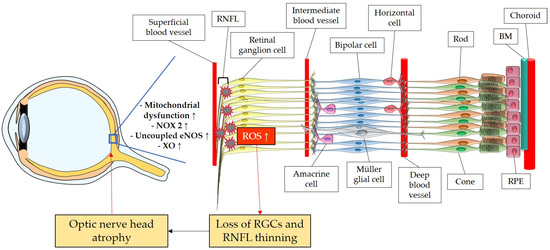
Figure 2. Model representing the ROS impact on the retina and on the optic nerve. ROS: reactive oxidative species; NOX2: NADPH oxidase type 2; XO: xanthine oxidase; eNOS: endothelial nitric oxide synthase; RGC: retinal ganglion cell; RNFL: retinal nerve fiber layer; RPE: retinal pigment epithelium; BM: Brunch’s membrane. Up arrows mean increase or upregulation.
References
- Riordan-Eva, P. Clinical assessment of optic nerve disorders. Eye Lond. 2004, 18, 1161–1168.
- Van Stavern, G.P.; Newman, N.J. Optic neuropathies. An overview. Ophthalmol. Clin. N. Am. 2001, 14, 61–71, viii.
- Sanz-Morello, B.; Ahmadi, H.; Vohra, R.; Saruhanian, S.; Freude, K.K.; Hamann, S.; Kolko, M. Oxidative Stress in Optic Neuropathies. Antioxidants 2021, 10, 1538.
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090.
- Stingl, J.V.; Wagner, F.M.; Liebezeit, S.; Baumgartner, R.; Spät, H.; Schuster, A.K.; Prokosch, V.; Grehn, F.; Hoffmann, E.M. Long-Term Efficacy and Safety of Modified Canaloplasty Versus Trabeculectomy in Open-Angle Glaucoma. Life 2023, 13, 516.
- Zhang, N.; Wang, J.; Li, Y.; Jiang, B. Prevalence of primary open angle glaucoma in the last 20 years: A meta-analysis and systematic review. Sci. Rep. 2021, 11, 13762.
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global causes of blindness and distance vision impairment 1990-2020: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e1221–e1234.
- Mascialino, B.; Leinonen, M.; Meier, T. Meta-analysis of the prevalence of Leber hereditary optic neuropathy mtDNA mutations in Europe. Eur. J. Ophthalmol. 2012, 22, 461–465.
- Puomila, A.; Hämäläinen, P.; Kivioja, S.; Savontaus, M.L.; Koivumäki, S.; Huoponen, K.; Nikoskelainen, E. Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland. Eur. J. Hum. Genet. 2007, 15, 1079–1089.
- Rosenberg, T.; Nørby, S.; Schwartz, M.; Saillard, J.; Magalhães, P.J.; Leroy, D.; Kann, E.C.; Duno, M. Prevalence and Genetics of Leber Hereditary Optic Neuropathy in the Danish Population. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1370–1375.
- Spruijt, L.; Kolbach, D.N.; de Coo, R.F.; Plomp, A.S.; Bauer, N.J.; Smeets, H.J.; de Die-Smulders, C.E. Influence of mutation type on clinical expression of Leber hereditary optic neuropathy. Am. J. Ophthalmol. 2006, 141, 676–682.
- Yu-Wai-Man, P.; Griffiths, P.G.; Brown, D.T.; Howell, N.; Turnbull, D.M.; Chinnery, P.F. The epidemiology of Leber hereditary optic neuropathy in the North East of England. Am. J. Hum. Genet. 2003, 72, 333–339.
- Watson, E.C.; Davis, R.L.; Ravishankar, S.; Copty, J.; Kummerfeld, S.; Sue, C.M. Low disease risk and penetrance in Leber hereditary optic neuropathy. Am. J. Hum. Genet. 2023, 110, 166–169.
- Mackey, D.A.; Ong, J.S.; MacGregor, S.; Whiteman, D.C.; Craig, J.E.; Lopez Sanchez, M.I.G.; Kearns, L.S.; Staffieri, S.E.; Clarke, L.; McGuinness, M.B.; et al. Is the disease risk and penetrance in Leber hereditary optic neuropathy actually low? Am. J. Hum. Genet. 2023, 110, 170–176.
- Li, K.J.; Semenov, D.; Turk, M.; Pope, J. A meta-analysis of the epidemiology of giant cell arteritis across time and space. Arthritis Res. Ther. 2021, 23, 82.
- Lee, J.Y.; Park, K.A.; Oh, S.Y. Prevalence and incidence of non-arteritic anterior ischaemic optic neuropathy in South Korea: A nationwide population-based study. Br. J. Ophthalmol. 2018, 102, 936–941.
- Toosy, A.T.; Mason, D.F.; Miller, D.H. Optic neuritis. Lancet Neurol. 2014, 13, 83–99.
- Braithwaite, T.; Subramanian, A.; Petzold, A.; Galloway, J.; Adderley, N.J.; Mollan, S.P.; Plant, G.T.; Nirantharakumar, K.; Denniston, A.K. Trends in Optic Neuritis Incidence and Prevalence in the UK and Association With Systemic and Neurologic Disease. JAMA Neurol. 2020, 77, 1514–1523.
- Rodriguez, M.; Siva, A.; Cross, S.A.; O’Brien, P.C.; Kurland, L.T. Optic neuritis: A population-based study in Olmsted County, Minnesota. Neurology 1995, 45, 244–250.
- Percy, A.K.; Nobrega, F.T.; Kurland, L.T. Optic neuritis and multiple sclerosis. An epidemiologic study. Arch. Ophthalmol. 1972, 87, 135–139.
- Cockerham, G.C.; Goodrich, G.L.; Weichel, E.D.; Orcutt, J.C.; Rizzo, J.F.; Bower, K.S.; Schuchard, R.A. Eye and visual function in traumatic brain injury. J. Rehabil. Res. Dev. 2009, 46, 811–818.
- Karimi, S.; Arabi, A.; Ansari, I.; Shahraki, T.; Safi, S. A Systematic Literature Review on Traumatic Optic Neuropathy. J. Ophthalmol. 2021, 2021, 5553885.
- Pirouzmand, F. Epidemiological trends of traumatic optic nerve injuries in the largest Canadian adult trauma center. J. Craniofac. Surg. 2012, 23, 516–520.
- Miller, N.R. Traumatic Optic Neuropathy. J. Neurol. Surg. B Skull Base 2021, 82, 107–115.
- Blandford, A.D.; Zhang, D.; Chundury, R.V.; Perry, J.D. Dysthyroid optic neuropathy: Update on pathogenesis, diagnosis, and management. Expert Rev. Ophthalmol. 2017, 12, 111–121.
- Neigel, J.M.; Rootman, J.; Belkin, R.I.; Nugent, R.A.; Drance, S.M.; Beattie, C.W.; Spinelli, J.A. Dysthyroid optic neuropathy. The crowded orbital apex syndrome. Ophthalmology 1988, 95, 1515–1521.
- Bartalena, L.; Piantanida, E.; Gallo, D.; Lai, A.; Tanda, M.L. Epidemiology, Natural History, Risk Factors, and Prevention of Graves’ Orbitopathy. Front. Endocrinol. 2020, 11, 615993.
- Kincaid, M.C.; Green, W.R. Ocular and orbital involvement in leukemia. Surv. Ophthalmol. 1983, 27, 211–232.
- Patel, L.; McNally, R.J.; Harrison, E.; Lloyd, I.C.; Clayton, P.E. Geographical distribution of optic nerve hypoplasia and septo-optic dysplasia in Northwest England. J. Pediatr. 2006, 148, 85–88.
- Tear Fahnehjelm, K.; Dahl, S.; Martin, L.; Ek, U. Optic nerve hypoplasia in children and adolescents; prevalence, ocular characteristics and behavioural problems. Acta Ophthalmol. 2014, 92, 563–570.
- Jefferis, J.M.; Hickman, S.J. Treatment and Outcomes in Nutritional Optic Neuropathy. Curr. Treat. Options Neurol. 2019, 21, 5.
- Roda, M.; di Geronimo, N.; Pellegrini, M.; Schiavi, C. Nutritional Optic Neuropathies: State of the Art and Emerging Evidences. Nutrients 2020, 12, 2653.
- Levin, L.A. Neuroprotection in Optic Neuropathy. Asia Pac. J. Ophthalmol. Phila. 2018, 7, 246–250.
- Casson, R.J.; Chidlow, G.; Wood, J.P.; Crowston, J.G.; Goldberg, I. Definition of glaucoma: Clinical and experimental concepts. Clin. Exp. Ophthalmol. 2012, 40, 341–349.
- Yu, D.-Y.; Cringle, S.J.; Balaratnasingam, C.; Morgan, W.H.; Yu, P.K.; Su, E.-N. Retinal ganglion cells: Energetics, compartmentation, axonal transport, cytoskeletons and vulnerability. Prog. Retin. Eye Res. 2013, 36, 217–246.
- Ruan, Y.; Jiang, S.; Musayeva, A.; Gericke, A. Oxidative Stress and Vascular Dysfunction in the Retina: Therapeutic Strategies. Antioxidants 2020, 9, 761.
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: Mechanisms and consequences. Prog. Retin. Eye Res. 2006, 25, 490–513.
- Mozaffarieh, M.; Flammer, J. New insights in the pathogenesis and treatment of normal tension glaucoma. Curr. Opin. Pharmacol. 2013, 13, 43–49.
- Aslan, M.; Dogan, S.; Kucuksayan, E. Oxidative stress and potential applications of free radical scavengers in glaucoma. Redox Rep. 2013, 18, 76–87.
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr. Opin. Pharmacol. 2013, 13, 12–15.
- Saccà, S.C.; Izzotti, A. Focus on molecular events in the anterior chamber leading to glaucoma. Cell. Mol. Life Sci. 2014, 71, 2197–2218.
- Langbøl, M.; Saruhanian, S.; Baskaran, T.; Tiedemann, D.; Mouhammad, Z.A.; Toft-Kehler, A.K.; Jun, B.; Vohra, R.; Bazan, N.G.; Kolko, M. Increased Antioxidant Capacity and Pro-Homeostatic Lipid Mediators in Ocular Hypertension—A Human Experimental Model. J. Clin. Med. 2020, 9, 2979.
- Masuda, T.; Shimazawa, M.; Hara, H. Retinal Diseases Associated with Oxidative Stress and the Effects of a Free Radical Scavenger (Edaravone). Oxid. Med. Cell. Longev. 2017, 2017, 9208489.
- Kang, E.Y.; Liu, P.K.; Wen, Y.T.; Quinn, P.M.J.; Levi, S.R.; Wang, N.K.; Tsai, R.K. Role of Oxidative Stress in Ocular Diseases Associated with Retinal Ganglion Cells Degeneration. Antioxidants 2021, 10, 1948.
- Harvey, J.P.; Sladen, P.E.; Yu-Wai-Man, P.; Cheetham, M.E. Induced Pluripotent Stem Cells for Inherited Optic Neuropathies-Disease Modeling and Therapeutic Development. J. Neuroophthalmol. 2022, 42, 35–44.
- Freddi, T.d.A.L.; Ottaiano, A.C. The Optic Nerve: Anatomy and Pathology. In Seminars in Ultrasound, CT and MRI; WB Saunders: Philadelphia, PA, USA, 2022; Volume 43, pp. 378–388.
- De Moraes, C.G. Anatomy of the visual pathways. J. Glaucoma 2013, 22 (Suppl. S5), S2–S7.
- Anderson, D.R. Vascular supply to the optic nerve of primates. Am. J. Ophthalmol. 1970, 70, 341–351.
- Linsenmeier, R.A.; Zhang, H.F. Retinal oxygen: From animals to humans. Prog. Retin. Eye Res. 2017, 58, 115–151.
- Arden, G.B.; Wolf, J.E.; Tsang, Y. Does dark adaptation exacerbate diabetic retinopathy?: Evidence and a linking hypothesis. Vis. Res. 1998, 38, 1723–1729.
- Selhorst, J.B.; Chen, Y. The optic nerve. Semin. Neurol. 2009, 29, 29–35.
- Prasad, S.; Galetta, S.L. Chapter 1—Anatomy and physiology of the afferent visual system. In Handbook of Clinical Neurology; Kennard, C., Leigh, R.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 102, pp. 3–19.
- Almazroa, A.; Burman, R.; Raahemifar, K.; Lakshminarayanan, V. Optic Disc and Optic Cup Segmentation Methodologies for Glaucoma Image Detection: A Survey. J. Ophthalmol. 2015, 2015, 180972.
- Dasgupta, S.; Mukherjee, R.; Dutta, K.; Sen, A. Deep learning based framework for automatic diagnosis of glaucoma based on analysis of focal notching in the optic nerve head. arXiv 2021, arXiv:2112.05748.
- Wilczek, M. The lamina cribrosa and its nature. Br. J. Ophthalmol. 1947, 31, 551–565.
- Emery, J.M.; Landis, D.; Paton, D.; Boniuk, M.; Craig, J.M. The lamina cribrosa in normal and glaucomatous human eyes. Trans. Am. Acad. Ophthalmol. Otolaryngol. 1974, 78, Op290–Op297.
- Quigley, H.A.; Hohman, R.M.; Addicks, E.M.; Massof, R.W.; Green, W.R. Morphologic changes in the lamina cribrosa correlated with neural loss in open-angle glaucoma. Am. J. Ophthalmol. 1983, 95, 673–691.
- Hayreh, S.S. Orbital vascular anatomy. Eye Lond. 2006, 20, 1130–1144.
- McCaa, C.S. The eye and visual nervous system: Anatomy, physiology and toxicology. Environ. Health Perspect. 1982, 44, 1–8.
- Juan, J.S.; Ana, I.R.; Rosa De, H.; Elena, S.-G.; Pilar, R.; José, A.F.-A.; Inés, L.-C.; Blanca, R.; Alberto, T.; José, M.R. Anatomy of the Human Optic Nerve: Structure and Function. In Optic Nerve; Felicia, M.F., Ed.; IntechOpen: Rijeka, Croatia, 2018; Chapter 2.
- Hattar, S.; Liao, H.W.; Takao, M.; Berson, D.M.; Yau, K.W. Melanopsin-containing retinal ganglion cells: Architecture, projections, and intrinsic photosensitivity. Science 2002, 295, 1065–1070.
- Wang, L.; Dong, J.; Cull, G.; Fortune, B.; Cioffi, G.A. Varicosities of intraretinal ganglion cell axons in human and nonhuman primates. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2–9.
- Bristow, E.A.; Griffiths, P.G.; Andrews, R.M.; Johnson, M.A.; Turnbull, D.M. The Distribution of Mitochondrial Activity in Relation to Optic Nerve Structure. Arch. Ophthalmol. 2002, 120, 791–796.
- Bartolucci, M.; Ravera, S.; Garbarino, G.; Ramoino, P.; Ferrando, S.; Calzia, D.; Candiani, S.; Morelli, A.; Panfoli, I. Functional Expression of Electron Transport Chain and FoF1-ATP Synthase in Optic Nerve Myelin Sheath. Neurochem. Res. 2015, 40, 2230–2241.
- Ravera, S.; Panfoli, I. Role of myelin sheath energy metabolism in neurodegenerative diseases. Neural Regen. Res. 2015, 10, 1570–1571.
- Bowley, M.P.; Cabral, H.; Rosene, D.L.; Peters, A. Age changes in myelinated nerve fibers of the cingulate bundle and corpus callosum in the rhesus monkey. J. Comp. Neurol. 2010, 518, 3046–3064.
- Depp, C.; Sun, T.; Sasmita, A.O.; Spieth, L.; Berghoff, S.A.; Nazarenko, T.; Overhoff, K.; Steixner-Kumar, A.A.; Subramanian, S.; Arinrad, S.; et al. Myelin dysfunction drives amyloid-β deposition in models of Alzheimer’s disease. Nature 2023, 618, 349–357.
- Wang, J.; He, X.; Meng, H.; Li, Y.; Dmitriev, P.; Tian, F.; Page, J.C.; Lu, Q.R.; He, Z. Robust Myelination of Regenerated Axons Induced by Combined Manipulations of GPR17 and Microglia. Neuron 2020, 108, 876–886.e4.
- Wong, K.A.; Benowitz, L.I. Retinal Ganglion Cell Survival and Axon Regeneration after Optic Nerve Injury: Role of Inflammation and Other Factors. Int. J. Mol. Sci. 2022, 23, 10179.
- Serasinghe, M.N.; Chipuk, J.E. Mitochondrial Fission in Human Diseases. Handb. Exp. Pharmacol. 2017, 240, 159–188.
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230.
- Walsh, C.; Barrow, S.; Voronina, S.; Chvanov, M.; Petersen, O.H.; Tepikin, A. Modulation of calcium signalling by mitochondria. Biochim. Biophys. Acta 2009, 1787, 1374–1382.
- Ward, P.S.; Thompson, C.B. Signaling in control of cell growth and metabolism. Cold Spring Harb. Perspect. Biol. 2012, 4, a006783.
- Elkholi, R.; Renault, T.T.; Serasinghe, M.N.; Chipuk, J.E. Putting the pieces together: How is the mitochondrial pathway of apoptosis regulated in cancer and chemotherapy? Cancer Metab. 2014, 2, 16.
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513.
- Shu, D.Y.; Chaudhary, S.; Cho, K.-S.; Lennikov, A.; Miller, W.P.; Thorn, D.C.; Yang, M.; McKay, T.B. Role of Oxidative Stress in Ocular Diseases: A Balancing Act. Metabolites 2023, 13, 187.
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344.
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89.
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15.
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31.
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763.
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84.
- Meza, C.A.; La Favor, J.D.; Kim, D.H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775.
- Taurone, S.; Ralli, M.; Artico, M.; Madia, V.N.; Scarpa, S.; Nottola, S.A.; Maconi, A.; Betti, M.; Familiari, P.; Nebbioso, M.; et al. Oxidative stress and visual system: A review. EXCLI J. 2022, 21, 544–553.
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837, 837a–837d.
- Kannenkeril, D.; Bosch, A.; Kolwelter, J.; Jung, S.; Striepe, K.; Ott, C.; Delles, C.; Schmieder, R.E. Dependency of flow-mediated vasodilatation from basal nitric oxide activity. Clin. Physiol. Funct. Imaging 2021, 41, 310–316.
- Tibballs, J. The role of nitric oxide (formerly endothelium-derived relaxing factor-EDRF) in vasodilatation and vasodilator therapy. Anaesth. Intensive Care 1993, 21, 759–773.
- Simonsen, U.; Rodriguez-Rodriguez, R.; Dalsgaard, T.; Buus, N.H.; Stankevicius, E. Novel approaches to improving endothelium-dependent nitric oxide-mediated vasodilatation. Pharmacol. Rep. 2009, 61, 105–115.
- Sanders, K.M.; Ward, S.M. Nitric oxide and its role as a non-adrenergic, non-cholinergic inhibitory neurotransmitter in the gastrointestinal tract. Br. J. Pharmacol. 2019, 176, 212–227.
- Rand, M.J.; Li, C.G. Nitric oxide as a neurotransmitter in peripheral nerves: Nature of transmitter and mechanism of transmission. Annu. Rev. Physiol. 1995, 57, 659–682.
- Vincent, S.R. Nitric oxide: A radical neurotransmitter in the central nervous system. Prog. Neurobiol. 1994, 42, 129–160.
- Falak, N.; Imran, Q.M.; Hussain, A.; Yun, B.W. Transcription Factors as the “Blitzkrieg” of Plant Defense: A Pragmatic View of Nitric Oxide’s Role in Gene Regulation. Int. J. Mol. Sci. 2021, 22, 522.
- Harari, O.; Liao, J.K. Inhibition of MHC II gene transcription by nitric oxide and antioxidants. Curr. Pharm. Des. 2004, 10, 893–898.
- Campbell, S.C.; Richardson, H.; Ferris, W.F.; Butler, C.S.; Macfarlane, W.M. Nitric oxide stimulates insulin gene transcription in pancreatic beta-cells. Biochem. Biophys. Res. Commun. 2007, 353, 1011–1016.
- Gunnett, C.A.; Lund, D.D.; Chu, Y.; Brooks, R.M., 2nd; Faraci, F.M.; Heistad, D.D. NO-dependent vasorelaxation is impaired after gene transfer of inducible NO-synthase. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1281–1287.
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424.
- Huie, R.E.; Padmaja, S. The reaction of no with superoxide. Free Radic. Res. Commun. 1993, 18, 195–199.
- Chronopoulos, P.; Manicam, C.; Zadeh, J.K.; Laspas, P.; Unkrig, J.C.; Göbel, M.L.; Musayeva, A.; Pfeiffer, N.; Oelze, M.; Daiber, A.; et al. Effects of Resveratrol on Vascular Function in Retinal Ischemia-Reperfusion Injury. Antioxidants 2023, 12, 853.
- Lei, Y.; Gao, Y.; Song, M.; Cao, W.; Sun, X. Peroxynitrite is a novel risk factor and treatment target of glaucoma. Nitric Oxide 2020, 99, 17–24.
- Cantó, A.; Olivar, T.; Romero, F.J.; Miranda, M. Nitrosative Stress in Retinal Pathologies: Review. Antioxidants 2019, 8, 543.
- Roos, D. Chronic Granulomatous Disease. Methods Mol. Biol. 2019, 1982, 531–542.
- Agita, A.; Alsagaff, M.T. Inflammation, Immunity, and Hypertension. Acta Med. Indones. 2017, 49, 158–165.
- Hsueh, Y.J.; Chen, Y.N.; Tsao, Y.T.; Cheng, C.M.; Wu, W.C.; Chen, H.C. The Pathomechanism, Antioxidant Biomarkers, and Treatment of Oxidative Stress-Related Eye Diseases. Int. J. Mol. Sci. 2022, 23, 1255.
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297.
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42.
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583.
- Smallwood, M.J.; Nissim, A.; Knight, A.R.; Whiteman, M.; Haigh, R.; Winyard, P.G. Oxidative stress in autoimmune rheumatic diseases. Free Radic. Biol. Med. 2018, 125, 3–14.
- Fleming, A.M.; Burrows, C.J. Chemistry of ROS-mediated oxidation to the guanine base in DNA and its biological consequences. Int. J. Radiat. Biol. 2022, 98, 452–460.
- Fleming, A.M.; Burrows, C.J. Interplay of Guanine Oxidation and G-Quadruplex Folding in Gene Promoters. J. Am. Chem. Soc. 2020, 142, 1115–1136.
- Jiang, M.; Zhao, X.M.; Jiang, Z.S.; Wang, G.X.; Zhang, D.W. Protein tyrosine nitration in atherosclerotic endothelial dysfunction. Clin. Chim. Acta 2022, 529, 34–41.
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 2020, 180, 968–983.e24.
- Radi, R.; Beckman, J.S.; Bush, K.M.; Freeman, B.A. Peroxynitrite-induced membrane lipid peroxidation: The cytotoxic potential of superoxide and nitric oxide. Arch. Biochem. Biophys. 1991, 288, 481–487.
- Hiona, A.; Leeuwenburgh, C. The role of mitochondrial DNA mutations in aging and sarcopenia: Implications for the mitochondrial vicious cycle theory of aging. Exp. Gerontol. 2008, 43, 24–33.
- Sanz, A.; Caro, P.; Gómez, J.; Barja, G. Testing the vicious cycle theory of mitochondrial ROS production: Effects of H2O2 and cumene hydroperoxide treatment on heart mitochondria. J. Bioenerg. Biomembr. 2006, 38, 121–127.
- Kaarniranta, K.; Pawlowska, E.; Szczepanska, J.; Jablkowska, A.; Blasiak, J. Role of Mitochondrial DNA Damage in ROS-Mediated Pathogenesis of Age-Related Macular Degeneration (AMD). Int. J. Mol. Sci. 2019, 20, 2374.
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell. Biol. 2019, 11, 1069–1082.
- Ali, S.S.; Ahsan, H.; Zia, M.K.; Siddiqui, T.; Khan, F.H. Understanding oxidants and antioxidants: Classical team with new players. J. Food Biochem. 2020, 44, e13145.
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71.
- Singhal, S.S.; Singh, S.P.; Singhal, P.; Horne, D.; Singhal, J.; Awasthi, S. Antioxidant role of glutathione S-transferases: 4-Hydroxynonenal, a key molecule in stress-mediated signaling. Toxicol. Appl. Pharmacol. 2015, 289, 361–370.
- Schipper, H.M.; Song, W.; Zukor, H.; Hascalovici, J.R.; Zeligman, D. Heme oxygenase-1 and neurodegeneration: Expanding frontiers of engagement. J. Neurochem. 2009, 110, 469–485.
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5.
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87.
- Dinkova-Kostova, A.T.; Talalay, P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol. Nutr. Food Res. 2008, 52 (Suppl. S1), S128–S138.
- Engwa, G.A. Free Radicals and the Role of Plant Phytochemicals as Antioxidants Against Oxidative Stress-Related Diseases. In Phytochemicals—Source of Antioxidants and Role in Disease Prevention; IntechOpen: London, UK, 2018.
- Yadav, A.; Mishra, P.C. Modeling the activity of glutathione as a hydroxyl radical scavenger considering its neutral non-zwitterionic form. J. Mol. Model. 2013, 19, 767–777.
- Stahl, W.; Sies, H. Antioxidant activity of carotenoids. Mol. Aspects Med. 2003, 24, 345–351.
- Gęgotek, A.; Skrzydlewska, E. Ascorbic acid as antioxidant. Vitam. Horm. 2023, 121, 247–270.
- Lee, G.Y.; Han, S.N. The Role of Vitamin E in Immunity. Nutrients 2018, 10, 1614.
- Rochette, L.; Ghibu, S.; Richard, C.; Zeller, M.; Cottin, Y.; Vergely, C. Direct and indirect antioxidant properties of α-lipoic acid. Mol. Nutr. Food Res. 2013, 57, 114–125.
- Xia, N.; Daiber, A.; Förstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1633–1646.
- Musayeva, A.; Unkrig, J.C.; Zhutdieva, M.B.; Manicam, C.; Ruan, Y.; Laspas, P.; Chronopoulos, P.; Göbel, M.L.; Pfeiffer, N.; Brochhausen, C.; et al. Betulinic Acid Protects from Ischemia-Reperfusion Injury in the Mouse Retina. Cells 2021, 10, 2440.
- Ito, Y.A.; Di Polo, A. Mitochondrial dynamics, transport, and quality control: A bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochondrion 2017, 36, 186–192.
- Joyal, J.S.; Gantner, M.L.; Smith, L.E.H. Retinal energy demands control vascular supply of the retina in development and disease: The role of neuronal lipid and glucose metabolism. Prog. Retin. Eye Res. 2018, 64, 131–156.
- Casson, R.J.; Chidlow, G.; Crowston, J.G.; Williams, P.A.; Wood, J.P.M. Retinal energy metabolism in health and glaucoma. Prog. Retin. Eye Res. 2021, 81, 100881.
- Wangsa-Wirawan, N.D.; Linsenmeier, R.A. Retinal Oxygen: Fundamental and Clinical Aspects. Arch. Ophthalmol. 2003, 121, 547–557.
- Cohen, L. Relationships between visual function and metabolism. In Biochemistry of the Eye; Graymore, C.N., Ed.; Academic Press: New York, NY, USA; London, UK, 1965; pp. 36–50.
- Anderson, B.; Saltzman, H.A. Retinal oxygen utilization measured by hyperbaric blackout. Arch. Ophthalmol. 1964, 72, 792–795.
- Ames, A., III. Energy requirements of CNS cells as related to their function and to their vulnerability to ischemia: A commentary based on studies on retina. Can. J. Physiol. Pharmacol. 1992, 70, S158–S164.
- Birk, M.; Baum, E.; Zadeh, J.K.; Manicam, C.; Pfeiffer, N.; Patzak, A.; Helmstadter, J.; Steven, S.; Kuntic, M.; Daiber, A.; et al. Angiotensin II Induces Oxidative Stress and Endothelial Dysfunction in Mouse Ophthalmic Arteries via Involvement of AT1 Receptors and NOX2. Antioxidants 2021, 10, 1238.
- Gericke, A.; Mann, C.; Zadeh, J.K.; Musayeva, A.; Wolff, I.; Wang, M.; Pfeiffer, N.; Daiber, A.; Li, H.; Xia, N.; et al. Elevated Intraocular Pressure Causes Abnormal Reactivity of Mouse Retinal Arterioles. Oxid. Med. Cell. Longev. 2019, 2019, 9736047.
- Wang, M.; Liu, H.; Xia, N.; Li, H.; van Beers, T.; Gericke, A.; Prokosch, V. Intraocular Pressure-Induced Endothelial Dysfunction of Retinal Blood Vessels Is Persistent, but Does Not Trigger Retinal Ganglion Cell Loss. Antioxidants 2022, 11, 1864.
- Zadeh, J.K.; Zhutdieva, M.B.; Laspas, P.; Yuksel, C.; Musayeva, A.; Pfeiffer, N.; Brochhausen, C.; Oelze, M.; Daiber, A.; Xia, N.; et al. Apolipoprotein E Deficiency Causes Endothelial Dysfunction in the Mouse Retina. Oxid. Med. Cell. Longev. 2019, 2019, 5181429.
- Zadeh, J.K.; Garcia-Bardon, A.; Hartmann, E.K.; Pfeiffer, N.; Omran, W.; Ludwig, M.; Patzak, A.; Xia, N.; Li, H.; Gericke, A. Short-Time Ocular Ischemia Induces Vascular Endothelial Dysfunction and Ganglion Cell Loss in the Pig Retina. Int. J. Mol. Sci. 2019, 20, 4685.
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551.
- Abramov, A.Y.; Scorziello, A.; Duchen, M.R. Three Distinct Mechanisms Generate Oxygen Free Radicals in Neurons and Contribute to Cell Death during Anoxia and Reoxygenation. J. Neurosci. 2007, 27, 1129–1138.
- Ono, T.; Tsuruta, R.; Fujita, M.; Aki, H.S.; Kutsuna, S.; Kawamura, Y.; Wakatsuki, J.; Aoki, T.; Kobayashi, C.; Kasaoka, S.; et al. Xanthine oxidase is one of the major sources of superoxide anion radicals in blood after reperfusion in rats with forebrain ischemia/reperfusion. Brain Res. 2009, 1305, 158–167.
- Dauth, A.; Breborowicz, A.; Ruan, Y.; Tang, Q.; Zadeh, J.K.; Bohm, E.W.; Pfeiffer, N.; Khedkar, P.H.; Patzak, A.; Vujacic-Mirski, K.; et al. Sulodexide Prevents Hyperglycemia-Induced Endothelial Dysfunction and Oxidative Stress in Porcine Retinal Arterioles. Antioxidants 2023, 12, 388.
- Giurdanella, G.; Lazzara, F.; Caporarello, N.; Lupo, G.; Anfuso, C.D.; Eandi, C.M.; Leggio, G.M.; Drago, F.; Bucolo, C.; Salomone, S. Sulodexide prevents activation of the PLA2/COX-2/VEGF inflammatory pathway in human retinal endothelial cells by blocking the effect of AGE/RAGE. Biochem. Pharmacol. 2017, 142, 145–154.
- Hein, T.W.; Xu, W.; Xu, X.; Kuo, L. Acute and Chronic Hyperglycemia Elicit JIP1/JNK-Mediated Endothelial Vasodilator Dysfunction of Retinal Arterioles. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4333–4340.
- Yan, S.D.; Schmidt, A.M.; Anderson, G.M.; Zhang, J.; Brett, J.; Zou, Y.S.; Pinsky, D.; Stern, D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J. Biol. Chem. 1994, 269, 9889–9897.
- Dehdashtian, E.; Mehrzadi, S.; Yousefi, B.; Hosseinzadeh, A.; Reiter, R.J.; Safa, M.; Ghaznavi, H.; Naseripour, M. Diabetic retinopathy pathogenesis and the ameliorating effects of melatonin; involvement of autophagy, inflammation and oxidative stress. Life Sci. 2018, 193, 20–33.
- Pang, B.; Li, M.; Song, J.; Li, Q.W.; Wang, J.; Di, S.; Tong, X.L.; Ni, Q. Luo Tong formula attenuates retinal inflammation in diabetic rats via inhibition of the p38MAPK/NF-κB pathway. Chin. Med. 2020, 15, 5.
- Lazzara, F.; Fidilio, A.; Platania, C.B.M.; Giurdanella, G.; Salomone, S.; Leggio, G.M.; Tarallo, V.; Cicatiello, V.; De Falco, S.; Eandi, C.M.; et al. Aflibercept regulates retinal inflammation elicited by high glucose via the PlGF/ERK pathway. Biochem. Pharmacol. 2019, 168, 341–351.
- Tarr, J.M.; Kaul, K.; Chopra, M.; Kohner, E.M.; Chibber, R. Pathophysiology of diabetic retinopathy. ISRN Ophthalmol. 2013, 2013, 343560.
- Lee, S.R.; An, E.J.; Kim, J.; Bae, Y.S. Function of NADPH Oxidases in Diabetic Nephropathy and Development of Nox Inhibitors. Biomol. Ther. Seoul 2020, 28, 25–33.
More