The research collects 65 unique structures, including spiro-biflavonoids, spiro-triflavonoids, spiro-tetraflavonoids, spiro-flavostilbenoids, and scillascillin-type homoisoflavonoids. Scillascillin-type homoisoflavonoids comprise spiro[bicyclo[4.2.0]octane-7,3′-chromane]-1(6),2,4-trien-4′-one, while the other spiro-flavonoids contain either 2H,2′H-3,3′-spirobi[benzofuran]-2-one or 2′H,3H-2,3′-spirobi[benzofuran]-3-one in the core of their structures. Spiro-flavonoids have been described in more than 40 species of eight families, including Asparagaceae, Cistaceae, Cupressaceae, Fabaceae, Pentaphylacaceae, Pinaceae, Thymelaeaceae, and Vitaceae. The possible biosynthetic pathways for each group of spiro-flavonoids are summarized in detail. Anti-inflammatory and anticancer activities are the most important biological activities of spiro-flavonoids, both in vitro and in vivo.
- spiro-flavonoids
- spiro-biflavonoids
- spiro-triflavonoids
- spiro-tetraflavonoids
- spiro-flavostilbenoids
- scillascillin-type homoisoflavonoids
- biosynthesis
- biological activity
- isolation
1. Introduction
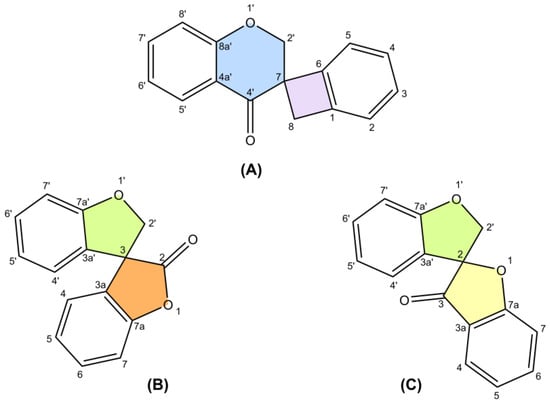
2. Biosynthesis of Spiro-Flavonoids
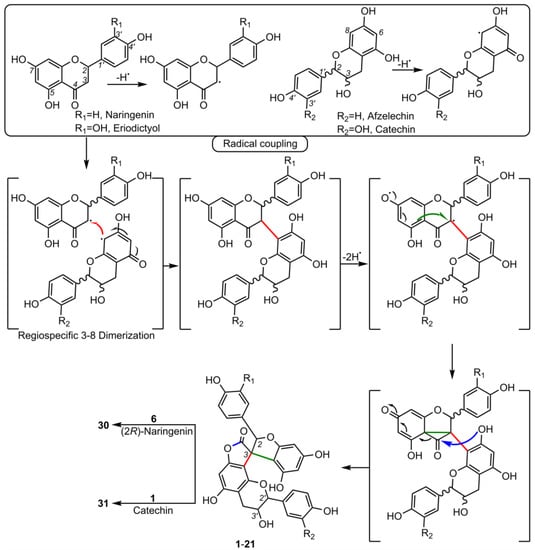
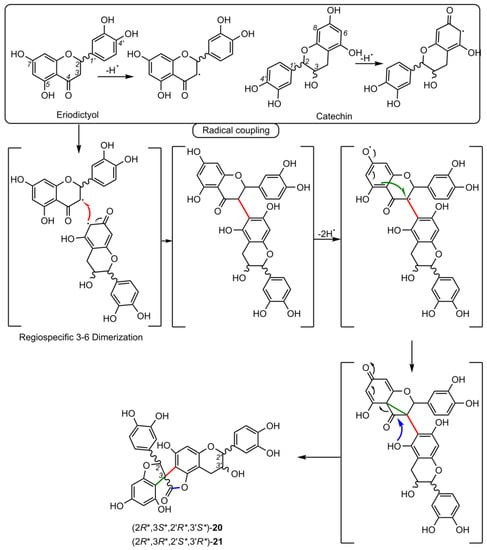
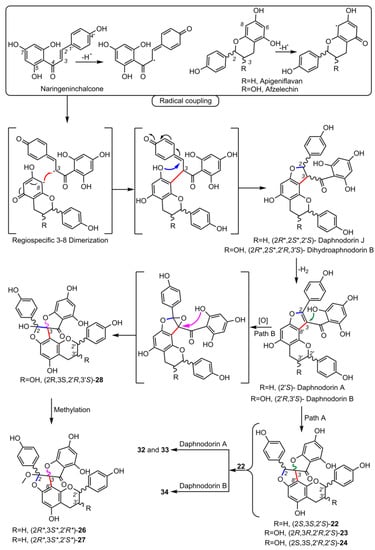
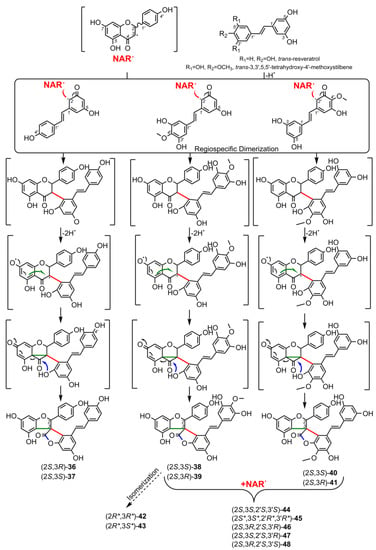
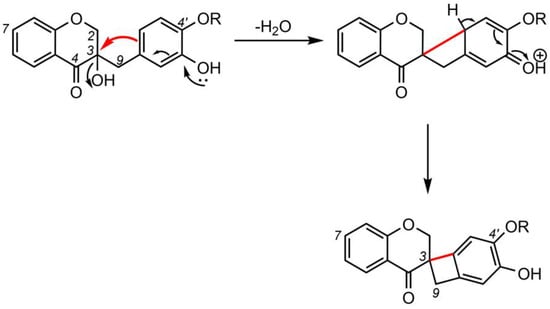
3. Biological Activities of Spiro-Flavonoids
3.1. Antioxidant Activity
The antioxidant capacity of the spiro-biflavonoid larixinol (1) originating from Larix decidua bark was evaluated in vitro using the DPPH assay (2,2-diphenyl-1-picrylhydrazyl) [50]. Compared to reference substances, larixinol was three times less active. Piacente et al. (2004) and Bassarello et al. (2007) tested the ability of 1 and yuccaols A-E (36–40), yuccaone A (29) from Yucca schidigera bark, and gloriosaols A-E (44–48) from Y. gloriosa root to scavenge the ABTS•+ radical (2,2′-azino-bis(3-ethylbenzothiozoline-6-sulfonic acid) diammonium salt) [51][52]. Their activity was expressed as Trolox Equivalent Antioxidant Capacity (TEAC) values and compared to quercetin (positive control) (Table 3). Comp. 44–46, and a mixture of gloriosaol D and E (47 and 48), showed the best antioxidant capacity in this study, much higher than quercetin [51]. Moderate ABTS•+ scavenging was observed for the dimeric spiro-flavonoids studied, with activity decreasing in the following order: yuccaol E, larixinol, yuccaol C, yuccaol D, and the weakest for yuccaol A and B, and yuccaone A (40, 1, 38, 39, 36, 37, 29, respectively) [52]. Compounds 36–40 in β-carotene/linoleic acid autoxidation assay showed significant activity, greater than the positive control—2,6-di-tert-butyl-4-methoxyphenol at 120 min [52]. Isolates 1, 29 were not as active as positive control in this assay [52]. The 15-LOX (lipoxygenase) inhibition assay was used to test the ability of 37 and 44 isolated from the bark of Y. schidigera to protect polyunsaturated acids from peroxidation [40]. Their antioxidant activity was higher (EC50 = 9.66 µg/mL for yuccaol B, EC50 = 12.34 µg/mL for gloriosaol A) compared to the positive control: ascorbic acid (EC50 = 21.52 µg/mL) [40]. Nishida et al. (2013) [53] investigated antioxidant activities in DPPH (Trolox, curcumin, and α-tocopherol as positive controls), hydrogen peroxide (H2O2; Trolox as a positive control), and nitric oxide (NO; Trolox and curcumin as positive controls) scavenging assays of scillascillin-type homoisoflavonoids 49, 51, 52, and 59 isolated from bulbs of Scilla scilloides. Isomuscomosin (52) tested at 500 μM showed more than 90% DPPH radical scavenging activity (EC50 = 22.9 μM; the other compounds tested were below 40%). In the H2O2 assay, scillascillin (49) and 2-hydroxy-scillascillin (51) showed activity below 40%, while isomuscomosin (52) and scillavone A (59) were highly active at 99.3% and 85.4%, respectively. In NO assays, the compounds tested were inactive (below 40% inhibition).3.2. Anti-Inflammatory Activity
Li et al. (2009) investigated the ability of spiro-biflavonoids (1–3) to reduce the level of NO production in macrophage cell line RAW 264.7, induced by lipopolysaccharide (LPS) [54]. Only larixinol showed NO inhibitory activity at 100 µg/mL with and IC50 value of 60.0 μg/mL [54]. Daphnodorin C and I (22, 24) and 2″-methoxy-daphnodorin C and 2″-methoxy-2-epi-daphnodorin C (26, 27) were tested for inhibitory activity against LPS-induced NO production in RAW 264.7 macrophages [55][56]. Only 26 and 27 showed statistically significant inhibitory activity of 32% and 58%, respectively, against an increase of NO at a concentration of 100 µg/mL (compared to 50% for the positive control, aminoguanidine at 25 µM). Compounds 22 and 24, isolated from the stems of Daphne kiusiana Miq., were investigated for their anti-inflammatory potential in the treatment of chronic obstructive pulmonary disease [57]. Comp. 22 most effectively suppressed the inflammatory response in both in vitro (phorbol 12-myristate 13-acetate)-stimulated human lung epithelial cells NCI-H292 and in an in vivo chronic obstructive pulmonary disease model, using mice exposed to cigarette smoke and LPS. Daphnodorin C (22) negatively affected the expression of inflammatory genes by inhibiting nuclear factor kappa light chain enhancer of activated B cells (NF-κB) and specific MAPK signaling pathways (mitogen-activated protein kinases) (JNK and p38) and suppressed reactive oxygen species (ROS) products in vitro and in vivo. Daphnodorin C at 20 mg/kg was comparable to the positive control roflumilast at 5 mg/kg [57]. A series of spiro-flavostilbenoids (yuccaols A-E, 36–40) from Y. schidigera bark was tested for anti-inflammatory activity using in vitro assays of COX-1, COX-2, and LTB4 (leukotriene B4) formation mediated by 5-LOX. Comp. 36 and 37, containing resveratrol as a stilbenic moiety, showed the highest inhibition against COX-1 and moderate inhibition against COX-2 [58]. Comp. 40, which has a THMS moiety in its structure, expressed a slightly lower COX-1 and COX-2 inhibitory activity than comp. 36 and 37. The investigated spiro-flavostilbenoids did not inhibit the formation of LTB4 [58]. In another in vitro study, yuccaols A-C (36–38) were tested on the J774.A1 murine macrophage cells activated with Escherichia coli LPS [59]. The anti-inflammatory effect was observed only for compounds 36 and 38 when they were added 1 h before LPS stimulation. Yuccaol C showed the highest activity. It significantly inhibited the iNOS (inducible nitric oxide synthase) protein and NO formation through NF-κB deactivation in a dose-dependent manner, while yuccaol A inhibited significantly only NO release at the highest concentration [59]. Spiro-flavostilbenoids 38–43 were evaluated for anti-inflammatory activity on the mouse macrophage cell line RAW 264.7 [60]. Cells were preincubated with the compounds at 100 µM for 1 h and then stimulated with LPS. Yuccalide B (42), yuccaol C (38), and yuccaol E (40), which had the same relative configuration, effectively suppressed the mRNA level of iNOS. Yuccaols C-E significantly reduced transcription of the inflammatory cytokines IL-6 and IL-1β [60]. Waller et al. (2013) tested a series of homoisoflavonoids (at 10 μM) isolated from Ledebouria socialis and L. ovatifolia, including socialinone (63) and C-2 acetylated forms of 2-hydroxy-7-O-methyl-scillascillin (50) for the inhibitory activity of COX-1 (SC-560 as a positive control) and COX-2 (DuP-607 as a positive control) [61]. These compounds were found to be inactive in COX-2 assay. Nishida et al. (2014) [62] investigated the anti-inflammatory properties of scillascillin-type homoisoflavonoids from bulbs of Scilla scilloides, 49, 51, 52, and 59, using lipoxygenase (nordihydroguaiaretic acid as a positive control) and hyaluronidase (tannic acid as a positive control) as a model of in vitro inflammation. However, the compounds were not active at any of the concentrations tested (500, 750, and 1000 μM). Furthermore, the authors tested the anti-inflammatory effect of scillascillin-type homoisoflavonoids on LPS-activated RAW 264.7 mouse macrophages. Compound 59 showed weak inhibitory activity at 10 μM, while 49, 51, 52, and 59 inhibited NO production level up to 50% at 50 μM. In another work [63], isomuscomosin (52) and compound 56 showed similar moderate anti-inflammatory activity in a microsomal fraction assay, while scillascillin (49) showed low activity (at concentrations of 250 μg/mL against indomethacin as a positive control). These compounds were found to be inactive in the COX-1 and COX-2 assays. Protosappanin D (65) and other constituents of Caesalpinia sappan were investigated using an in vitro assay with the J774.1 cell line [64]. Their inhibitory effects on NO and prostaglandin E2 (PGE2), as well as their suppressive effects on the expression of tumor necrosis factor-α (TNF-α), IL-6, COX-2, and iNOS mRNA level, were evaluated. As a result, 65 inhibited both NO (IC50 = 9.6 μM) and PGE2 (IC50 = 7.8 μM) production. It showed the strongest suppression of TNF-α (IC50 = 14.2 μM), IL-6 (IC50 = 3.0 μM), COX-2 (IC50 = 21.4 μM), and iNOS mRNA expression (IC50 = 13.2 μM) among the substances tested.3.3. Neuroprotective Activity
Spiro-flavostilbenoids (36–41, 44, 46–48) and spiro-biflavonoids (6, 7) from the bark of Y. schidigera were tested in vitro for their inhibitory activity against cholinesterases [7][65]. Acetylcholinesterase (AChE) from electric eel and butyrylcholinesterase (BChE) from horse serum were used in a modified Ellman spectrophotometric assay [7][65]. Compared to galantamine (positive control), the compounds tested showed moderate or weak inhibition of AChE. However, yuccaol B (37) and gloriosaol A (44) were the most potent inhibitors of BChE, with IC50 lower than the positive control (81.3 μM for 37, 64.9 μM for 44, and 124.0 μM for galantamine) [7]. Among yuccaols, the (2S,3S) diastereoisomers showed the highest anticholinesterase activity. The molecular interactions between the most active compounds (37, 44) and human AChE/BChE were studied in silico. These compounds interacted similarly with the peripheral anionic site of AChE and were located deep in the catalytic site of BChE [7]. The neuroprotective effect of yuccaol B and gloriosaol A was also confirmed in vivo using adult zebrafish models [40]. The Y-maze test was used to assess the function of spatial working memory function, while the novel tank diving test was used to measure anxiety in Danio rerio. Spiro-flavostilbenoids 37 and 44 at doses of 1, 3, and 5 µg/L attenuated scopolamine-induced amnesia and anxiety, and improved precognitive and anxiolytic activities, to the level of the control group (untreated with scopolamine) [40].3.4. Anticancer and Antitumor Activity
Spiro-biflavonoids abiesinols A-F (1, 4, 12–15), derived from the bark of Abies sachalinensis, were screened in vitro for antitumor-initiating activity, and then one of the most active compounds (12) was tested in vivo for a skin cancer chemopreventive effect [66]. Their inhibitory effect on the activation of NOR 1 ((±)-(E)-methyl-2-[(E)-hydroxyimino]-5-nitro-6-methoxy-hex-3-enamide), a donor of nitric oxide, was evaluated in Chang human liver cells, using curcumin as a reference compound. The inhibitory activity of all compounds tested was similar to that of curcumin; only abiesinols E (1) and F (4), which have fewer OH groups in their structure than abiesinols A-D, were slightly less active. A two-step skin carcinogenesis assay was performed using specific-pathogen-free female ICR mice (6 weeks old). Abiesinol A (12) was administered orally (0.0025% of body weight) for 2 weeks; skin carcinogenesis was induced with a single dose of peroxynitrite (ONOO-) and promoted with 12-O-tetradecanoylphorbol-13-acetate applied topically twice a week for 20 weeks [66]. Compound 12 showed significant antitumor-initiating activity in the in vivo test, the percentage of papilloma bearing mice was reduced to 33% at week 11, tumor formation was delayed by 2 weeks, and at the end of the experiment the average number of papillomas per mouse was reduced by a factor of 2, compared to the control group (no abiesinol A treatment) [66]. In contrast, the spiro-biflavonoids 3-epi-larixinol (2) and fragranols B and C (8, 9), as well as the spiro-triflavonoid fragranol A (30), were not active against the human pancreatic cancer cell line PANC-1 in an in vitro assay. Compounds 2, 8, 9, and 30 did not kill tumor cells at the maximum tested amount of 100 μM [34][35]. Spiro-biflavonoid genkwanol A (23), isolated from the aerial part of Fumana procumbens (Dunal) Gren. & Godr., was evaluated in vitro for its anticancer activity against A549 (adenocarcinomic human basal alveolar epithelial cell line), MCF-7 (human breast cancer cell line), HeLa (human cervical cancer cell line), and BEAS-2B (human bronchial epithelial cell line) cells using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) assay [67]. Compound 23, at the highest concentration tested (20 μg/mL), was active only against A549 cells, showing a 16.8% inhibition of cell viability [67]. Similarly, 23 had a moderate antimitotic effect, compared to the positive controls of colchicine and vinblastine in the microtubule polymerization bioassay in vitro [68]. In another work, the affinity of 23, daphnodorin I (24), 2″-hydroxygenkwanol A (28), and 4′-methylgenkwanol A (25) to Hsp90 protein, one of the most promising targets for anticancer therapy, was tested in vitro [69]. All spiro-biflavonoids interacted with the immobilized protein, but comp. 28 was the most efficient [69]. Spiro-flavostilbenoids 36–38 inhibited Kaposi sarcoma (KS) cell proliferation, migration, and synthesis of the inflammatory mediator PAF (platelet-activating factor) in vitro [70]. Yuccaol C (38) was the most active, and it completely blocked the growth of the VEGF (vascular endothelial growth factor)-induced cells, more efficiently attenuated p38 and p42/44 MAP kinase signaling pathways activated by VEGF, and completely suppressed cell migration of KS cells after the PAF treatment. Moreover, yuccaols A-C inactivated VEGF-induced PAF biosynthesis through acetyl transferase blockade and enhanced PAF degradation through lysophospholipid transacetylase activation [70]. The cytostatic and pro-apoptotic activities of gloriosaols A-C (44–46) were tested in vitro against different cancer cell lines: MCF7 (breast carcinoma), HepG2 (hepatoblastoma), U937 (monocytic leukemia), Molt4 (lymphoblastic leukemia), and Jurkat (T-cell leukemia) [71]. Inhibition of cancer cell growth was observed after 24 h treatment with increasing concentrations of 44–46. These stereoisomers showed different antiproliferative potentials: 46 had the lowest EC50 values, and the best effect was observed towards the U937 cell line, followed by 44 and 45 [71]. At the highest doses of 10–25 µM, gloriosaol C (46) induced apoptosis in the most sensitive cell line, U937, and tended to switch to necrosis at doses above 30 µM. However, the cytotoxic (proapoptotic) effect of 46 against human peripheral blood mononuclear cells was distinctly weaker. Gloriosaol C-induced apoptosis at doses > 10 µM caused mitochondrial depolarization and cytochrome c release in U937 cells [71]. The 44–46 tests altered the intracellular redox balance by increasing ROS (reactive oxygen species) release in U937 cancer cells exposed to these compounds for 1 h, and reducing ROS levels in cells simultaneously exposed to a pro-oxidant agent; that is, t-butyl hydroperoxide. The best pro-oxidant and antioxidant effect was observed for 46 at doses higher than 10 µM. This is one of the mechanisms by which these stereoisomers (44–46) were able to induce apoptosis in U937 cells. [71]. Schwikkard et al. (2019) [72] reported the antiproliferative activity of several structurally diverse homoisoflavonoids (including 49, 52, 53, 63, and 64) from Ledebouria ovatifolia and Chionodoxa luciliae Boiss. against endothelial tumor cells (human retinal microvascular endothelial cells) and ocular tumor cells (uveal melanoma 92-1 and retinoblastoma Y79). The authors indicate that the activity of homoisoflavonoids tested was generally related to the substitution pattern of their A and B rings. However, the presence of the spiro-ring rendered the tested scillascillin-type homoisoflavonoids completely inactive, regardless of the substituents in the A and B rings. Likewise, Matsuo et al. (2014) [73] evaluated (3R)-5,7-dihydroxy-6-methyl-3-(3′-hydroxy-4′-methoxybenzyl)chroman-4-one and compound 57 (isolated from Bessera elegans), which differed only in the presence of the 3-spiro-cyclobutene ring (formed by the coupling of C-3 and C-2′ carbons), for cytotoxicity against human HL-60 promyelocytic leukemia cells and normal human diploid TIG-3 fibroblasts. The homoisoflavonoid without the cyclobutene ring showed a potent tumor-selective cytotoxic activity against HL-60 cells, while 57 was noncytotoxic (and noncytotoxic against TIG-3). On the other hand, Chinthala et al. (2014) [74] performed an in vitro anticancer assay using scillascillin (49) isolated from Ledebouria hyderabadensis against the human cancer cell lines MCF-7 (breast cancer) and DU-145 (prostate cancer). Scillascillin showed significant activity (IC50 of 9.59 ug/mL and 11.32 μg/mL, respectively). Doxorubicin was used as a positive control. Its IC50 against the tested cell lines was 1.86 and 13.71 μg/mL, respectively.3.5. Cytotoxicity/Mutagenicity
The lack of cytotoxicity of spiro-biflavonoid larixinol (1) at a dose of 100 µg/mL was confirmed in the RAW264.7 macrophages using the MTT assay [54]. No cytotoxicity of daphnodorin C (22) and daphnodorin I (24) at concentrations of 2.5–20 µM was detected against NCI-H292 human lung epithelial cells using the Cell Counting Kit-8 assay [57]. Spiro-flavostilbenoids 36–38 (yuccaols A-C) showed no toxicity against J774.A1 murine monocyte/macrophage cells at doses of 0.1–100 µM in an MTT test [59]. These compounds, at doses of 10–500 µg, confirmed their non-toxicity and demonstrated their non-mutagenicity in Salmonella typhimurium strains TA97, TA98, TA100, and TA102 tested by the microsome test (Ames test) [75].3.6. Antiplatelet Activity
Sakuma et al. (1998) [76] tested spiro-biflavonoid daphnodorin C (22) isolated from Daphne odora roots for the activities of 12-lipoxygenase (12-LOX) and cyclooxygenase (COX). 12-LOX converts arachidonic acid to 12-hydroperoxy-5,8,10,14-eicosatetraenoic acid, which is subsequently reduced to 12-hydroxy-5,8,10,14-eicosatetraenoic acid (12-HETE). 12-HETE has been reported to induce platelet aggregation. COX, on the other hand, produces prostaglandin endoperoxides. These are converted by thromboxane synthase to 12-hydroxy-5,8,10-heptadecatrienoic acid (HHT). Daphnodorin C (22) caused 34.1% inhibition of 12-HETE formation and 26.6% inhibition of HHT at a concentration of 100 μM. This makes 22 a dual inhibitor of 12-LOX and COX in platelets. In vitro, the spiro-flavostilbenoids 36 and 38 yuccaols A and C showed an inhibitory effect on platelet aggregation induced by thrombin and ADP (adenosine diphosphate) in a dose-dependent manner. Pig blood platelets pretreated with the compounds 36 and 38 at their highest concentration (25 μg/mL) caused a suppression of thrombin-induced aggregation by about 50% [77].3.7. Antidiabetic Activity
A series of spiro-biflavonoid diastereoisomers (1–4, 10, 11) isolated from the stem bark of Glyptostrobus pensilis were screened in vitro for their ability to inhibit human protein tyrosine phosphatase 1B (PTP1B). This enzyme negatively regulates insulin signaling or epidermal growth factor pathways [78]. Spiropensilisol A (10) showed the best activity, inhibiting the enzyme to the same extent as a positive control (oleanolic acid) with an IC50 =3.3 µM. The remaining compounds tested were also active, with an IC50 ≤ 17.1 µM. Using a molecular modeling approach, compound 10 was shown to interact with the catalytic site of the PTP1B enzyme [78]. Daphnodorins C and I (22, 24) and spiro-tetraflavonoids edgechrins A, B, and D (32, 34, 33) isolated from Edgeworthia chrysantha Lindl. were tested for α-glucosidase inhibitory activity, using p-nitrophenyl α-D-glucoside as substrate [79]. All compounds significantly inhibited α-glucosidase, but dimeric compounds 32–34 had a stronger activity than 22 and 24 (IC50 1.09, 0.96, 2.13 versus 4.00 and 19.0, respectively, with 73.6 μM for the positive control, acarbose).3.8. Antibacterial, Antifungal, and Antiviral Activity
Inamori et al. (1987) [80] tested the antifungal activity of spiro-biflavonoid daphnodorin C (22) against pathogens including Pyricularia oryzae, Rhizoctonia solani, Phytophora infestans, Botrytis cinerea, Puccinia recondita, and Erysiphe graminis, and its insecticidal activity (against Spodoptera litura, Nilaparvata lugens, Callosobruchus chinensis, and Tetranychus urticae). It showed antifungal activity only against P. oryzae associated with Oriza sativa as a plant host. The leaves of rice plant were incubated with daphnodorin C for 3 d before being inoculated with fungal spores for 5 d. The protective value of 22 was 89–90% at a concentration of 200–500 ppm. The insecticidal activity was negligible. Genkwanol A (23) caused an in vitro morphological deformation of the phytopathogenic fungus responsible for rice blast—P. oryzae. Compared to antifungal controls, that is, griseofulvin and nocodazole, 23 had lower minimum morphological deformation concentration [68]. Daphnodorin C (22), isolated from the bark of Dahpne odora Thunb., showed moderate inhibition of HIV-1 replication in MT-4 cells [81]. Compared to a positive control, 2′,3′-dideoxycytidine- 5′-triphosphate, comp. 22 had a weak inhibitory effect on HIV-1 reverse transcriptase. The authors suggest that daphnodorin C has anti-HIV-1 effects through inhibition of the early stage of viral replication [81]. Genkwanol A (23), derived from the root of Wikstroemia indica (L.) C.A.Mey., showed moderate activity against human immunodeficiency virus type 1 (HIV-1) in T4 lymphocytes (CEM cell line) [68]. Antibacterial activity of scillascillin-type homoisoflavonoids 49, 52, and 56 isolated from Drimiopsis maculata Lindl. & Paxton, and Eucomis schijffii Reyneke, was screened against Staphylococcus aureus, using the bioautographic and microplate assays [82]. Significant inhibitory activity was obtained for scillascillin (49), with a minimum inhibitory concentration (MIC) value of 0.50 mM (neomycin, positive control, had MIC = 0.0025 mM), while compound 52 (isomuscomosin) exhibited bacteriostatic activity with a bacteriostatic concentration value of 1.97 mM.3.9. Phytotoxic Activity
Spiro-biflavonoid genkwanol A (23), isolated from the roots of Stellera chamaejasme L., showed strong phytotoxic activity against Arabidopsis thaliana seedlings, although only at relatively high concentrations (200 μg/mL), with an IC50 of 74.8 μg/mL, by inhibiting root growth [83]. This was confirmed by measuring the level of endogenous auxin in the root tip of the transgenic A. thaliana DR5::GUS line. This level was significantly reduced by 23 as a result of inhibition of auxin transport. At the same time, comp. 23 was not secreted into the soil by the roots of S. chamaejasme. Therefore, it is unlikely that it is responsible for the allelopathic effect exerted by this plant in nature.3.10. Other Activities
Angiotensin II plays a key role in regulating blood pressure, and Takai et al. (1999) [84] tested the inhibition of human chymase-dependent angiotensin II-forming activity by daphnodorin A, daphnodorin B, and daphnodorin C (22) isolated from D. odora. The compound did not inhibit chymase-generated angiotensin II formation, but also did not affect the formation of angiotensin-converting enzyme-dependent angiotensin II and, unlike daphnodorin A, did not inhibit purified human tryptase. The spiro-biflavonoids genkwanol A (23) and 2″-hydroxygenkwanol A (28) were tested in vitro for poly(ADP-ribose) polymerase 1 (PARP-1) inhibitory activity [85]. PARP-1 is an enzyme involved in the pathogenesis of cancer, inflammation, diabetes, and neurodegenerative diseases. Compound 28 binds efficiently to the PARP-1 protein catalytic domain in the nicotine binding pocket. 2″-Hydroxygenkwanol A strongly inhibited PARP-1 activity at submicromolar concentrations at a level comparable to the positive control—3-aminobenzamide [85]. Fusi et al. (2010) [86] investigated the vasorelaxant effects of scillascillin-type homoisoflavonoids 49, 52, and 56 obtained from D. maculata and E. schijffii using rat aortic ring preparations. The authors reported that both 60 mM K+ (K60) and phenylephrine-induced tonic contractions were inhibited, in a concentration-dependent manner, by all homoisoflavonoids tested. Compound 56 was found to be the most effective vasorelaxing agent. This was in part due to the activation of soluble guanylyl cyclase. Sasaki et al. (2010) [87] investigated the vasorelaxant activity of protosappanin D (65) isolated from C. sappan on the rat aorta and mesenteric artery. The authors reported that 65 exhibited vasorelaxing activity on both phenylephrine-preconstricted blood vessels and that its activity was independent of the aortic endothelium and dependent on the mesenteric artery endothelium. The involvement of NO and prostaglandin as endothelium-derived relaxing factors was demonstrated in further experiments with NG-nitro-L-arginine and indomethacin.4. Conclusions
Spiro-flavonoids, due to the presence of an unusual structural element such as spiro-carbon, are attracting increasing interest because of their chemical and biological properties. A total of 65 spiro-flavonoid structures which were monomeric, as well as bi-, tri-, and tetrameric, belonging to several groups differing in the type of polyphenolic units and the way they are combined, were isolated and characterized. Spiro-biflavonoids were the most abundant group, most frequently isolated from the families Pinaceae, Thymelaeaceae, Cupressaceae, and Pentaphylacaceae. In turn, the richest source of spiro-compounds (thirty-four structures) was the Asparagaceae family, from which all known scillascillin-type homoisoflavonoids and all spiro-flavostilbenoids were derived. Most of the oligomeric spiro-flavonoids were isolated from woody plant parts (twigs, bark, and roots), while monomeric scillascillin-type homoisoflavonoids were obtained from bulbs. Methods used to isolate them mainly included classical extraction by maceration at room temperature using pure organic solvents of relatively different polarity and their mixtures, most often with water. The subsequent separation steps also included classical separation techniques based on the difference in solubility in two immiscible liquids (liquid-liquid extraction) as well as the use of column liquid chromatography in normal and reversed-phase systems and gel filtration.
The relative and absolute configurations of the complex structures of spiro-flavonoids, frequently containing multiple chiral carbons (including spiro-carbons), have been determined by a number of spectroscopic techniques, including nuclear magnetic resonance (NMR), electronic circular dichroism (ECD), X-ray diffraction (XRD), and chemical methods using chiral derivatizing agents. NMR and XRD are the methods most commonly used to determine their relative configurations, although an increasing number of cases of the use of quantum mechanical (QM) calculations (e.g., modified DP4+ probability method) are reported in the literature. Empirical methods have been used to assign absolute configuration, including the comparison of Cotton effects between known and newly described compounds. However, this method seems to be far from sufficient for structures with more than one chirality center, so it is necessary to systematically and correctly use QM techniques to predict ECD spectra using time-dependent density-functional theory calculations.
The potential health benefits of spiro-flavonoids have been summarized and the available results indicate significant anti-inflammatory, neuroprotective, antitumor/anticancer, and antidiabetic properties in vitro and in vivo of some spiro-biflavonoids and spiro-flavostilbenoids. On the other hand, scillascillin-type homoisoflavonoids showed good vasorelaxant activity. Therefore, future research should focus on these aspects of their activity.
References
- Moss, G.P. Extension and Revision of the Nomenclature for Spiro Compounds. Pure Appl. Chem. 1999, 71, 531–558.
- Ding, A.; Meazza, M.; Guo, H.; Yang, J.W.; Rios, R. New Development in the Enantioselective Synthesis of Spiro Compounds. Chem. Soc. Rev. 2018, 47, 5946–5996.
- Rios, R. Enantioselective Methodologies for the Synthesis of Spiro Compounds. Chem. Soc. Rev. 2012, 41, 1060–1074.
- Hiesinger, K.; Dar’in, D.; Proschak, E.; Krasavin, M. Spirocyclic Scaffolds in Medicinal Chemistry. J. Med. Chem. 2021, 64, 150–183.
- Kouno, I.; Komori, T.; Kawasaki, T. Zur Struktur Der Neuen Typen Homo-Isoflavanone Aus Bulben von Scilla scilloides Druce. Tetrahedron Lett. 1973, 14, 4569–4572.
- Fedorova, T.E.; Ivanova, S.Z.; Fedorov, S.V.; Babkin, V.A. Larisinol, a New Spirobiflavonoid from Larix gmelinii Bark. Chem. Nat. Compd. 2007, 43, 208–209.
- Pecio, Ł.; Alilou, M.; Kozachok, S.; Orhan, I.E.; Eren, G.; Şenol Deniz, F.S.; Stuppner, H.; Oleszek, W. Absolute Configuration of Spiro-Flavostilbenoids from Yucca schidigera Roezl Ex Ortgies: First Indication of (2R)-Naringenin as the Key Building Block. Phytochemistry 2023, 207, 113584.
- Grotewold, E. The Genetics and Biochemistry of Floral Pigments. Annu. Rev. Plant Biol. 2006, 57, 761–780.
- Tropf, S.; Lanz, T.; Rensing, S.A.; Schröder, J.; Schröder, G. Evidence That Stilbene Synthases Have Developed from Chalcone Synthases Several Times in the Course of Evolution. J. Mol. Evol. 1994, 38, 610–618.
- Forkmann, G.; Heller, W. Confirm. In Comprehensive Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 1999; pp. 713–748.
- Bednar, R.A.; Hadcock, J.R. Purification and Characterization of Chalcone Isomerase from Soybeans. J. Biol. Chem. 1988, 263, 9582–9588.
- Shirley, B.W.; Kubasek, W.L.; Storz, G.; Bruggemann, E.; Koornneef, M.; Ausubel, F.M.; Goodman, H.M. Analysis of Arabidopsis Mutants Deficient in Flavonoid Biosynthesis. Plant J. 1995, 8, 659–671.
- Cahn, R.S.; Ingold, C.K.; Prelog, V. The Specification of Asymmetric Configuration in Organic Chemistry. Experientia 1956, 12, 81–94.
- Zeb, N.; Rashid, M.H.; Mubarak, M.Q.E.; Ghafoor, S.; de Visser, S.P. Flavonol Biosynthesis by Nonheme Iron Dioxygenases: A Computational Study into the Structure and Mechanism. J. Inorg. Biochem. 2019, 198, 110728.
- Lukačin, R.; Wellmann, F.; Britsch, L.; Martens, S.; Matern, U. Flavonol Synthase from Citrus unshiu Is a Bifunctional Dioxygenase. Phytochemistry 2003, 62, 287–292.
- Britsch, L.; Grisebach, H. Purification and Characterization of (2S)-Flavanone 3-hydroxylase from Petunia hybrida. Eur. J. Biochem. 1986, 156, 569–577.
- Ferrer, J.-L.; Austin, M.B.; Stewart, C.; Noel, J.P. Structure and Function of Enzymes Involved in the Biosynthesis of Phenylpropanoids. Plant Physiol. Biochem. 2008, 46, 356–370.
- Hanhineva, K.; Kokko, H.; Siljanen, H.; Rogachev, I.; Aharoni, A.; Kärenlampi, S.O. Stilbene Synthase Gene Transfer Caused Alterations in the Phenylpropanoid Metabolism of Transgenic Strawberry (Fragaria×ananassa). J. Exp. Bot. 2009, 60, 2093–2106.
- Jeandet, P.; Vannozzi, A.; Sobarzo-Sánchez, E.; Uddin, M.S.; Bru, R.; Martínez-Márquez, A.; Clément, C.; Cordelier, S.; Manayi, A.; Nabavi, S.F.; et al. Phytostilbenes as Agrochemicals: Biosynthesis, Bioactivity, Metabolic Engineering and Biotechnology. Nat. Prod. Rep. 2021, 38, 1282–1329.
- Liu, Z.; Zhuang, C.; Sheng, S.; Shao, L.; Zhao, W.; Zhao, S. Overexpression of a Resveratrol Synthase Gene (PcRS) from Polygonum Cuspidatum in Transgenic Arabidopsis Causes the Accumulation of Trans-Piceid with Antifungal Activity. Plant Cell Rep. 2011, 30, 2027–2036.
- Nicoletti, I.; De Rossi, A.; Giovinazzo, G.; Corradini, D. Identification and Quantification of Stilbenes in Fruits of Transgenic Tomato Plants (Lycopersicon esculentum Mill.) by Reversed Phase HPLC with Photodiode Array and Mass Spectrometry Detection. J. Agric. Food Chem. 2007, 55, 3304–3311.
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The Grapevine Genome Sequence Suggests Ancestral Hexaploidization in Major Angiosperm Phyla. Nature 2007, 449, 463–467.
- González-Barrio, R.; Beltrán, D.; Cantos, E.; Gil, M.I.; Espín, J.C.; Tomás-Barberán, F.A. Comparison of Ozone and UV-C Treatments on the Postharvest Stilbenoid Monomer, Dimer, and Trimer Induction in Var. ‘Superior′ White Table Grapes. J. Agric. Food Chem. 2006, 54, 4222–4228.
- Pezet, R.; Perret, C.; Jean-Denis, J.B.; Tabacchi, R.; Gindro, K.; Viret, O. δ-Viniferin, a Resveratrol Dehydrodimer: One of the Major Stilbenes Synthesized by Stressed Grapevine Leaves. J. Agric. Food Chem. 2003, 51, 5488–5492.
- Timperio, A.M.; D’Alessandro, A.; Fagioni, M.; Magro, P.; Zolla, L. Production of the Phytoalexins Trans-Resveratrol and Delta-Viniferin in Two Economy-Relevant Grape Cultivars upon Infection with Botrytis cinerea in Field Conditions. Plant Physiol. Biochem. 2012, 50, 65–71.
- Calderón, A.A.; Zapata, J.M.; Pedreño, M.A.; Muñoz, R.; Barceló, A.R. Levels of 4-Hydroxystilbene-Oxidizing Isoperoxidases Related to Constitutive Disease Resistance in in Vitro-Cultured Grapevine. Plant Cell. Tissue Organ Cult. 1992, 29, 63–70.
- Langcake, P.; Pryce, R.J. A New Class of Phytoalexins from Grapevines. Experientia 1977, 33, 151–152.
- Oleszek, W.; Sitek, M.; Stochmal, A.; Piacente, S.; Pizza, C.; Cheeke, P. Resveratrol and Other Phenolics from the Bark of Yucca schidigera Roezl. J. Agric. Food Chem. 2001, 49, 747–752.
- Shen, Z.; Haslam, E.; Falshaw, C.P.; Begley, M.J. Procyanidins and Polyphenols of Larix gmelini Bark. Phytochemistry 1986, 25, 2629–2635.
- Zhou, B.; Alania, Y.; Reis, M.C.; McAlpine, J.B.; Bedran-Russo, A.K.; Pauli, G.F.; Chen, S.-N. Rare A-Type, Spiro-Type, and Highly Oligomeric Proanthocyanidins from Pinus massoniana. Org. Lett. 2020, 22, 5304–5308.
- Gorham, J.; Coughlan, S.J. Inhibition of Photosynthesis by Stilbenoids. Phytochemistry 1980, 19, 2059–2064.
- Sangha, A.K.; Parks, J.M.; Standaert, R.F.; Ziebell, A.; Davis, M.; Smith, J.C. Radical Coupling Reactions in Lignin Synthesis: A Density Functional Theory Study. J. Phys. Chem. B 2012, 116, 4760–4768.
- Elder, T.; Rencoret, J.; del Río, J.C.; Kim, H.; Ralph, J. Radical Coupling Reactions of Hydroxystilbene Glucosides and Coniferyl Alcohol: A Density Functional Theory Study. Front. Plant Sci. 2021, 12, 319.
- Omar, A.M.; Sun, S.; Kim, M.J.; Tawila, A.M.; Dibwe, D.F.; Phrutivorapongkul, A.; Toyooka, N.; Awale, S. Fragranol A: A New Class of Spiro-Triflavanoid Hybrid with an Unprecedented Carbon Skeleton from Anneslea fragrans. Tetrahedron Lett. 2020, 61, 152099.
- Omar, A.M.; Sun, S.; Kim, M.J.; Tawila, A.M.; Dibwe, D.F.; Phrutivorapongkul, A.; Toyooka, N.; Awale, S. Highly Oxygenated Spiro-Biflavanoids from Anneslea fragrans Twigs. Phytochem. Lett. 2020, 40, 21–25.
- Wada, S.; Hitomi, T.; Tanaka, R. Phenolic Compounds Isolated from the Bark of Abies sachalinensis. Helv. Chim. Acta 2009, 92, 1610–1620.
- Givens, R.S.; Heger, D.; Hellrung, B.; Kamdzhilov, Y.; Mac, M.; Conrad, P.G.; Cope, E.; Lee, J.I.; Mata-Segreda, J.F.; Schowen, R.L.; et al. The Photo-Favorskii Reaction of p-Hydroxyphenacyl Compounds Is Initiated by Water-Assisted, Adiabatic Extrusion of a Triplet Biradical. J. Am. Chem. Soc. 2008, 130, 3307–3309.
- Baldwin, J.E. Rules for Ring Closure. J. Chem. Soc. Chem. Commun. 1976, 734–736.
- Baldwin, J.E.; Thomas, R.C.; Kruse, L.I.; Silberman, L. Rules for Ring Closure: Ring Formation by Conjugate Addition of Oxygen Nucleophiles. J. Org. Chem. 1977, 42, 3846–3852.
- Pecio, Ł.; Kozachok, S.; Brinza, I.; Stefan Boiangiu, R.; Hritcu, L.; Mircea, C.; Flavia Burlec, A.; Cioanca, O.; Hancianu, M.; Wronikowska-Denysiuk, O.; et al. Neuroprotective Effect of Yucca schidigera Roezl Ex Ortgies Bark Phenolic Fractions, Yuccaol B and Gloriosaol A on Scopolamine-Induced Memory Deficits in Zebrafish. Molecules 2022, 27, 3692.
- Castelli, M.V.; López, S.N. Homoisoflavonoids: Occurrence, Biosynthesis, and Biological Activity. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2017; Volume 54, pp. 315–354.
- Dewick, P.M. Biosynthesis of the 3-Benzylchroman-4-One Eucomin. J. Chem. Soc. Chem. Commun. 1973, 438–439.
- Dewick, P.M. Biosynthesis of the 3-Benzylchroman-4-One Eucomin in Eucomis bicolor. Phytochemistry 1975, 14, 983–988.
- Zheng, Y.; Tice, C.M.; Singh, S.B. The Use of Spirocyclic Scaffolds in Drug Discovery. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682.
- Zheng, Y.J.; Tice, C.M. The Utilization of Spirocyclic Scaffolds in Novel Drug Discovery. Expert Opin. Drug Discov. 2016, 11, 831–834.
- Benabdallah, M.; Talhi, O.; Nouali, F.; Choukchou-Braham, N.; Bachari, K.; Silva, A.M.S. Advances in Spirocyclic Hybrids: Chemistry and Medicinal Actions. Curr. Med. Chem. 2018, 25, 3748–3767.
- Acosta-Quiroga, K.; Rojas-Peña, C.; Nerio, L.S.; Gutiérrez, M.; Polo-Cuadrado, E. Spirocyclic Derivatives as Antioxidants: A Review. RSC Adv. 2021, 11, 21926–21954.
- Yang, J.; Wang, Y.; Guan, W.; Su, W.; Li, G.; Zhang, S.; Yao, H. Spiral Molecules with Antimalarial Activities: A Review. Eur. J. Med. Chem. 2022, 237, 114361.
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756.
- Baldan, V.; Sut, S.; Faggian, M.; Dalla Gassa, E.; Ferrari, S.; De Nadai, G.; Francescato, S.; Baratto, G.; Dall’Acqua, S. Larix decidua Bark as a Source of Phytoconstituents: An LC-MS Study. Molecules 2017, 22, 1974.
- Bassarello, C.; Bifulco, G.; Montoro, P.; Skhirtladze, A.; Benidze, M.; Kemertelidze, E.; Pizza, C.; Piacente, S. Yucca gloriosa: A Source of Phenolic Derivatives with Strong Antioxidant Activity. J. Agric. Food Chem. 2007, 55, 6636–6642.
- Piacente, S.; Montoro, P.; Oleszek, W.; Pizza, C. Yucca schidigera Bark: Phenolic Constituents and Antioxidant Activity. J. Nat. Prod. 2004, 67, 882–885.
- Nishida, Y.; Wada, K.; Toyohisa, D.; Tanaka, T.; Ono, M.; Yasuda, S. Homoisoflavones as the Antioxidants Responsible from Bulbs of Scilla scilloides. Nat. Prod. Res. 2013, 27, 2360–2362.
- Li, Y.-L.; Yang, X.-W.; Li, S.-M.; Tang, J.; Tian, J.-M.; Peng, X.-Y.; Huang, D.-S.; Zhang, W.-D. Two New Spirobiflavonoids from Abies Chensiensis with Moderate NO Production Inhibitory Activity. Planta Med. 2009, 75, 1534–1537.
- Liang, S.; Tian, J.-M.; Feng, Y.; Liu, X.-H.; Xiong, Z.; Zhang, W.-D. Flavonoids from Daphne aurantiaca and Their Inhibitory Activities against Nitric Oxide Production. Chem. Pharm. Bull. 2011, 59, 653–656.
- Liang, S.; Tang, J.; Shen, Y.-H.; Jin, H.-Z.; Tian, J.-M.; Wu, Z.-J.; Zhang, W.-D.; Yan, S.-K. Biflavonoids from Daphne feddei and Their Inhibitory Activities against Nitric Oxide Production. Chem. Pharm. Bull. 2008, 56, 1729–1731.
- Ryu, H.W.; Lee, J.W.; Kim, M.O.; Lee, R.W.; Kang, M.J.; Kim, S.M.; Min, J.H.; Oh, E.S.; Song, Y.N.; Jung, S.; et al. Daphnodorin C Isolated from the Stems of Daphne kiusiana Miquel Attenuates Airway Inflammation in a Mouse Model of Chronic Obstructive Pulmonary Disease. Phytomedicine 2022, 96, 153848.
- Wenzig, E.M.; Oleszek, W.; Stochmal, A.; Kunert, O.; Bauer, R. Influence of Phenolic Constituents from Yucca schidigera Bark on Arachidonate Metabolism in Vitro. J. Agric. Food Chem. 2008, 56, 8885–8890.
- Marzocco, S.; Piacente, S.; Pizza, C.; Oleszek, W.; Stochmal, A.; Pinto, A.; Sorrentino, R.; Autore, G. Inhibition of Inducible Nitric Oxide Synthase Expression by Yuccaol C from Yucca schidigera Roezl. Life Sci. 2004, 75, 1491–1501.
- Nakashima, K.; Abe, N.; Oyama, M.; Inoue, M. Yuccalides A–C, Three New Phenolic Compounds with Spiro-Structures from the Roots of Yucca gloriosa. Fitoterapia 2016, 111, 154–159.
- Waller, C.P.; Thumser, A.E.; Langat, M.K.; Crouch, N.R.; Mulholland, D.A. COX-2 Inhibitory Activity of Homoisoflavanones and Xanthones from the Bulbs of the Southern African Ledebouria socialis and Ledebouria ovatifolia (Hyacinthaceae: Hyacinthoideae). Phytochemistry 2013, 95, 284–290.
- Nishida, Y.; Sugahara, S.; Wada, K.; Toyohisa, D.; Tanaka, T.; Ono, M.; Yasuda, S. Inhibitory Effects of the Ethyl Acetate Extract from Bulbs of Scilla scilloides on Lipoxygenase and Hyaluronidase Activities. Pharm. Biol. 2014, 52, 1351–1357.
- Du Toit, K.; Elgorashi, E.E.; Malan, S.F.; Drewes, S.E.; Van Staden, J.; Crouch, N.R.; Mulholland, D.A. Anti-Inflammatory Activity and QSAR Studies of Compounds Isolated from Hyacinthaceae Species and Tachiadenus longiflorus Griseb. (Gentianaceae). Bioorg. Med. Chem. 2005, 13, 2561–2568.
- Washiyama, M.; Sasaki, Y.; Hosokawa, T.; Nagumo, S. Anti-Inflammatory Constituents of Sappan Lignum. Biol. Pharm. Bull. 2009, 32, 941–944.
- Pecio, Ł.; Alilou, M.; Kozachok, S.; Orhan, I.E.; Eren, G.; Deniz, F.S.S.; Stuppner, H.; Oleszek, W. Yuccalechins A-C from the Yucca schidigera Roezl Ex Ortgies Bark: Elucidation of the Relative and Absolute Configurations of Three New Spirobiflavonoids and Their Cholinesterase Inhibitory Activities. Molecules 2019, 24, 4162.
- Wada, S.-I.; Hitomi, T.; Tokuda, H.; Tanaka, R. Anti-Tumor-Initiating Effects of Spiro-Biflavonoids from Abies sachalinensis. Chem. Biodivers. 2010, 7, 2303–2308.
- Emerce, E.; Gürbüz, P.; Doğan, Ş.D.; Kadioglu, E.; Süntar, I. Cytotoxic Activity-Guided Isolation Studies on Fumana procumbens (Dunal) Gren. & Godr. Rec. Nat. Prod. 2019, 13, 189–198.
- Hu, K.; Kobayashi, H.; Dong, A.; Iwasaki, S.; Yao, X. Antifungal, Antimitotic and Anti-HIV-1 Agents from the Roots of Wikstroemia indica. Planta Med. 2000, 66, 564–567.
- Malafronte, N.; Vassallo, A.; Dal Piaz, F.; Bader, A.; Braca, A.; De Tommasi, N. Biflavonoids from Daphne linearifolia Hart. Phytochem. Lett. 2012, 5, 621–625.
- Balestrieri, C.; Felice, F.; Piacente, S.; Pizza, C.; Montoro, P.; Oleszek, W.; Visciano, V.; Balestrieri, M.L. Relative Effects of Phenolic Constituents from Yucca schidigera Roezl. Bark on Kaposi’s Sarcoma Cell Proliferation, Migration, and PAF Synthesis. Biochem. Pharmacol. 2006, 71, 1479–1487.
- Nigro, P.; Bloise, E.; Turco, M.C.; Skhirtladze, A.; Montoro, P.; Pizza, C.; Piacente, S.; Belisario, M.A. Antiproliferative and Pro-Apoptotic Activity of Novel Phenolic Derivatives of Resveratrol. Life Sci. 2007, 81, 873–883.
- Schwikkard, S.; Whitmore, H.; Sishtla, K.; Sulaiman, R.S.; Shetty, T.; Basavarajappa, H.D.; Waller, C.; Alqahtani, A.; Frankemoelle, L.; Chapman, A.; et al. The Antiangiogenic Activity of Naturally Occurring and Synthetic Homoisoflavonoids from the Hyacinthaceae (Sensu APGII). J. Nat. Prod. 2019, 82, 1227–1239.
- Matsuo, Y.; Kurihara, R.; Akagi, N.; Mimaki, Y. Two New Homoisoflavonoids from the Bulbs of Bessera elegans. Nat. Prod. Commun. 2014, 9, 1725–1727.
- Chinthala, Y.; Chinde, S.; Kumar, A.N.; Srinivas, K.V.N.S.; Kumar, J.K.; Sastry, K.P.; Grover, P.; Ramana, M.V. Anticancer Active Homoisoflavone from the Underground Bulbs of Ledebouria hyderabadensis. Pharmacogn. Res. 2014, 6, 303–305.
- Czeczot, H.; Podsiad, M.; Skrzycki, M.; Stochmal, A.; Oleszek, W. Evaluation of the Mutagenic Activity of Phenolics from the Bark of Yucca schidigera Roezl. Acta Pol. Pharm. 2003, 60, 357–362.
- Sakuma, S.; Fujimoto, Y.; Tsunomori, M.; Tagano, S.; Nishida, H.; Baba, K.; Fujita, T. Effects of Daphnodorin A, B and C, New Flavans Isolated from Traditional Chinese Medicine, on the 12. Lipoxygenase and Cyclooxygenase Metabolism of Arachidonic Acid in Rabbit Platelets. Prostaglandins, Leukot. Essent. Fat. Acids 1998, 58, 143–146.
- Olas, B.; Wachowicz, B.; Stochmal, A.; Oleszek, W. Anti-Platelet Effects of Different Phenolic Compounds from Yucca schidigera Roezl. Bark. Platelets 2002, 13, 167–173.
- Xiong, J.; Hu, C.L.; Wang, P.P.; Gao, D.D.; Huang, F.; Li, J.; Hu, J.F. Spirobiflavonoid Stereoisomers from the Endangered Conifer Glyptostrobus pensilis and Their Protein Tyrosine Phosphatase 1B Inhibitory Activity. Bioorg. Med. Chem. Lett. 2020, 30, 126943.
- Zhou, T.; Zhang, S.W.; Liu, S.S.; Cong, H.J.; Xuan, L.J. Daphnodorin Dimers from Edgeworthia chrysantha with α-Glucosidase Inhibitory Activity. Phytochem. Lett. 2010, 3, 242–247.
- Inamori, Y.; Takeuchi, K.; Baba, K.; Kozawa, M. Antifungal and Insecticidal Activities of Daphnodorins A, B and C. Chem. Pharm. Bull. 1987, 35, 3931–3934.
- Yusa, K.; Oh-hara, T.; Tsukahara, S.; Baba, K.; Taniguchi, M.; Kozawa, M.; Takeuchi, S.; Hara, H.; Tsuruo, T. Inhibition of Human Immunodeficiency Virus Type 1 (HIV-1) Replication by Daphnodorins. Antivir. Res. 1994, 25, 57–66.
- Du Toit, K.; Elgorashi, E.E.; Malan, S.F.; Mulholland, D.A.; Drewes, S.E.; Van Staden, J. Antibacterial Activity and QSAR of Homoisoflavanones Isolated from Six Hyacinthaceae Species. S. Afr. J. Bot. 2007, 73, 236–241.
- Yan, Z.; Guo, H.; Yang, J.; Liu, Q.; Jin, H.; Xu, R.; Cui, H.; Qin, B. Phytotoxic Flavonoids from Roots of Stellera chamaejasme L. (Thymelaeaceae). Phytochemistry 2014, 106, 61–68.
- Takai, S.; Sakaguchi, M.; Jin, D.; Baba, K.; Miyazaki, M. Effects of Daphnodorin A, Daphnodorin B and Daphnodorin C on Human Chymase-Dependent Angiotensin II Formation. Life Sci. 1999, 64, 1889–1896.
- Dal Piaz, F.; Ferro, P.; Vassallo, A.; Vasaturo, M.; Forte, G.; Chini, M.G.; Bifulco, G.; Tosco, A.; De Tommasi, N. Identification and Mechanism of Action Analysis of the New PARP-1 Inhibitor 2″-Hydroxygenkwanol A. Biochim. Biophys. Acta-Gen. Subj. 2015, 1850, 1806–1814.
- Fusi, F.; Ferrara, A.; Koorbanally, C.; Crouch, N.R.; Mulholland, D.A.; Sgaragli, G. Vascular Myorelaxing Activity of Isolates from South African Hyacinthaceae Partly Mediated by Activation of Soluble Guanylyl Cyclase in Rat Aortic Ring Preparations. J. Pharm. Pharmacol. 2010, 60, 489–497.
- Sasaki, Y.; Suzuki, M.; Matsumoto, T.; Hosokawa, T.; Kobayashi, T.; Kamata, K.; Nagumo, S. Vasorelaxant Activity of Sappan Lignum Constituents and Extracts on Rat Aorta and Mesenteric Artery. Biol. Pharm. Bull. 2010, 33, 1555–1560.