Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lindsay Dong and Version 1 by Beatriz Martinez-Delgado.
Non-alcoholic fatty liver disease (NAFLD) is a type of steatosis commonly associated with obesity, dyslipidemia, hypertension, and diabetes. Other diseases such as inherited alpha-1 antitrypsin deficiency (AATD) have also been related to the development of liver steatosis. The primary reasons leading to hepatic lipid deposits can be genetic and epigenetic, and the outcomes range from benign steatosis to liver failure, as well as to extrahepatic diseases. Progressive hepatocellular damage and dysregulated systemic immune responses can affect extrahepatic organs, specifically the heart and lungs.
- NAFLD
- AATD
- cardiovascular disease
- lipids
- meta-inflammation
1. Non-Alcoholic Fatty Liver Disease (NAFLD)
Non-alcoholic fatty liver disease (NAFLD) is a common liver condition characterized by an excess of lipid accumulation in hepatocytes (steatosis), which is present in about 25% of the adult population [1]. This term includes a range of liver diseases from benign steatosis to cirrhosis, passing through steatohepatitis (NASH) to hepatocellular carcinoma (HCC) [2]. There are different environmental or genetic risk factors that can lead to NAFLD [3], including insulin resistance and obesity. MAFLD (metabolic associated fatty liver disease) has been proposed as a new name that is expected to better mirror the heterogeneities and similarities between NAFLD and metabolic syndrome [4,5][4][5]; however, some controversies remain regarding this new name [6].
The pathology typically begins with an altered lipid homeostasis, the intracellular increment of fats followed by an uncontrolled inflammatory response, which can eventually lead to cirrhosis and/or to HCC [7]. Initially, most of the NAFLD patients are asymptomatic and blood markers typically do not reflect liver impairment [8]. The progression to NASH is associated with liver inflammation usually followed by fibrosis, whereas in some cases, the development of liver failure requires liver transplantation. However, cardiovascular diseases (CVD) are among the main causes of death among NAFLD patients [9].
It is widely accepted that free fatty acids act as primary triggers of NAFLD, although there are other factors implicated in disease progression such as dietary habits, obesity, insulin resistance, intestinal microbiota, or epigenetic factors [10]. Patients with NASH typically have high levels of blood endotoxins, suggesting that bacterial endotoxins play a role in NASH pathogenesis [11,12][11][12].
Steatosis is defined by the presence of lipid droplets (LDs) in the cytosol of more than 5% of hepatocytes, which is a consequence of altered lipid metabolism when fatty acid obtention exceeds fatty acid removal [14][13]. Lipid droplets are dynamic organelles composed of neutral lipids, mainly triglycerides and cholesterol esters [15][14], which act as energy storage but also as protectors against the deleterious effects of free fatty acids [16][15]. LDs are increasingly recognized as having important non-pathological roles in cell signalling and function. The properties of LDs are highly regulated by proteins coating the surface of LDs to control lipid trafficking and flux [17][16]. LDs also play roles in endoplasmic reticulum (ER) stress response, protein storage and degradation, and in infection and immunity [18][17].

2. Alpha-1 Antitrypsin Deficiency (AATD)
Inherited alpha-1 antitrypsin deficiency (AATD) is a rare monogenic disorder (ORPHA 60) mainly related to lung and/or liver diseases, but also to neutrophilic panniculitis or systemic vasculitis [20][18]. AATD is characterized by low levels of circulating alpha-1 antitrypsin (AAT), an acute phase glycoprotein encoded by the SERPINA1 gene, in which more than 120 allelic variants have been described [21][19]. Some mutations in the SERPINA1 gene have no clinical relevance and are considered as normal variants or M alleles; however, deficient alleles, typically resulting from point mutations or small deletions, are related to low levels or functional activity of AAT, and mild to severe clinical manifestations. Among the deficient alleles, the most clinically relevant and best recognized is the Z allele (Glu342Lys), originating from a point mutation in exon 5 [22][20]. According to current data, the homozygosity in the Z allele is present in about 96% of AATD patients, whereas the remaining 4% are heterozygous carriers or contain other rare alleles [23][21]. AAT is primarily synthetized by hepatocytes (about 80%) and acts not only as a main inhibitor of neutrophil elastase and proteinase-3 [24[22][23],25], but also as a modulator of caspase activity and apoptosis, as an antioxidant, and/or as a broad immunomodulatory protein [26,27][24][25]. The complex tertiary structure of AAT makes it extremely vulnerable to conformational changes, as it happens in the Z allele where a change in just one amino acid triggers AAT polymerization. As mentioned above, AATD mainly affects the liver and lungs; hepatic manifestations are due to AAT intrahepatic polymer accumulation and cytotoxicity [28][26], whereas lung pathologies are due to low circulating levels, mostly polymeric forms of AAT resulting in an insufficient inhibition of neutrophil proteases [29][27].3. Meta-Inflammation in NAFLD and AATD
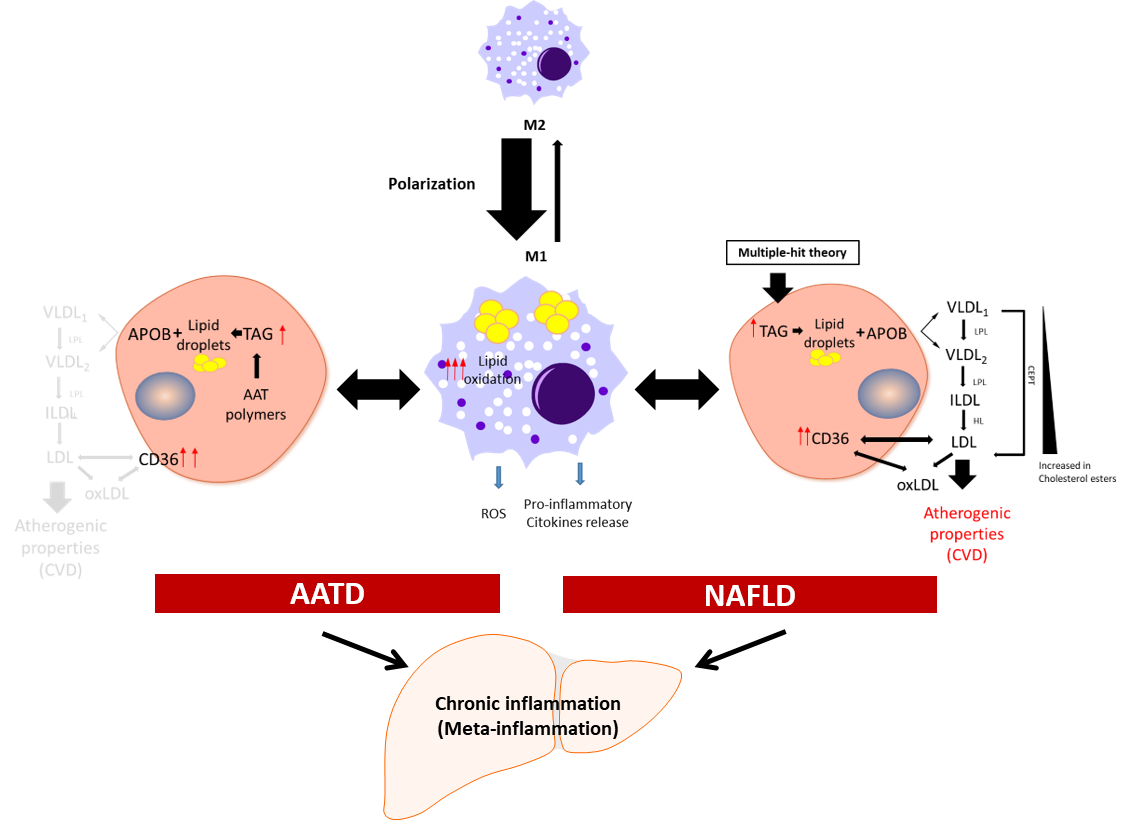
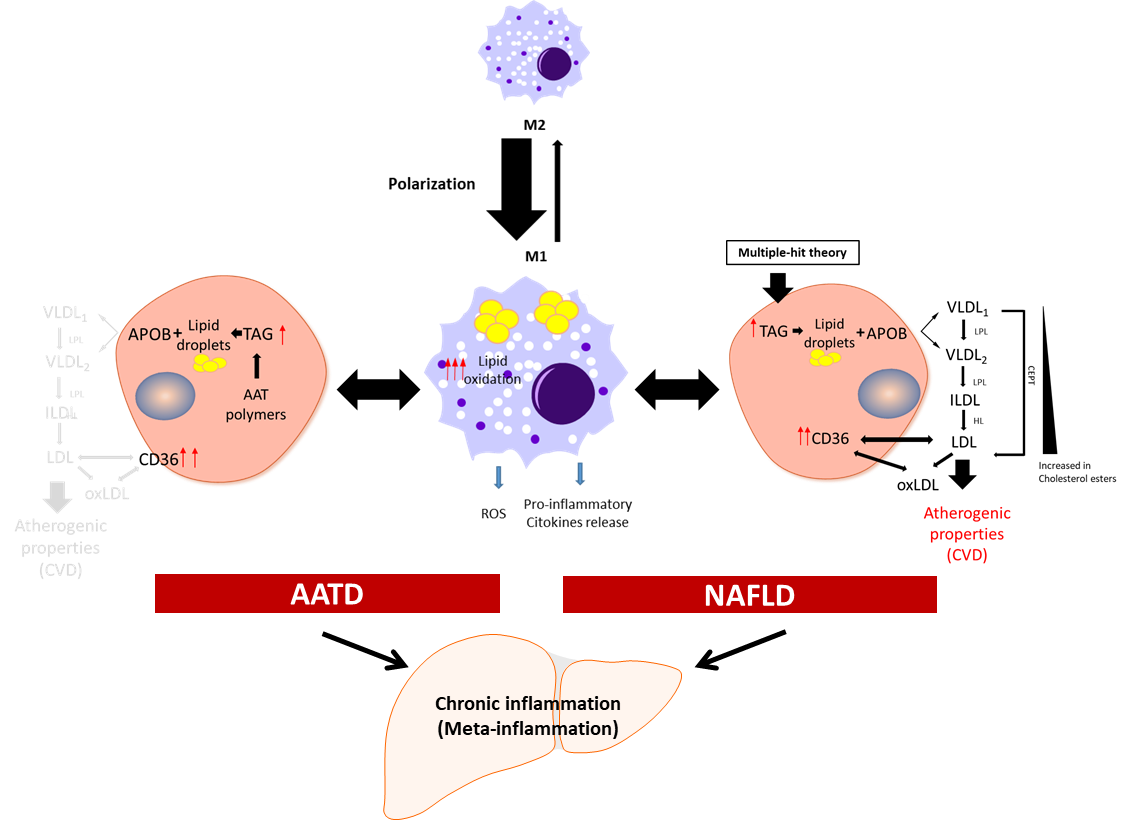
Meta-inflammation is defined as a low-grade chronic inflammation associated with metabolic syndrome [39][28]. Most scientists agree that meta-inflammation, as a component of immune system, links chronic inflammatory diseases and obesity [40][29]. In this scenario, adipose tissue macrophages can react to high concentrations of fatty acids and initiate signalling pathways promoting monocyte mobilization and differentiation into macrophages, which further contribute to the inflammatory response [41,42][30][31]. Macrophages derived from hematopoietic progenitors are involved in homeostatic and pathogenic processes. In adult tissues, the functions of macrophages are dependent on the microenvironment, and thus macrophages can acquire a proinflammatory (M1) or an anti-inflammatory (anti-fibrotic) (M2) phenotype [43,44][32][33]. Bone-marrow monocyte-derived macrophages can also acquire a pro-inflammatory phenotype and contribute to inflammation [45][34]. Because of lipid accumulation in NAFLD, not only is macrophage polarization altered in favour of the M1 phenotype, but macrophages also undergo metabolic reprogramming leading to increased fatty acid intake and worsen steatosis [46][35]. Activated liver Kupffer cells release pro-inflammatory cytokines, which in turn activate hepatic stellate cells, hepatocytes, or endothelial cells [47[36][37],48], promoting monocyte infiltration and boosting macrophage population. Furthermore, fat accumulation in Kupffer cells leads to oxidative stress and structural changes in the plasmatic and mitochondrial membranes, while in the context of AATD, due to AAT protein accumulation in the ER, this also leads to activation of the unfolded protein response [14][13]. An increase in free fatty acids intensifies lipid oxidation, mainly in the mitochondria and peroxisomes, as well as free-radical production, which can lead to mitochondrial damage and fragmentation [49,50][38][39]. On the other hand, ER stress induced by misfolded proteins triggering the unfolded protein response elicits p53 expression, mitochondrial cytochrome c release, and apoptosis [51][40]. Hence, liver Kupffer cells can contribute not only to the sustained meta-inflammation, but also to the progression of NAFLD (Figure 1).
Figure 1. Schematic presentation of the development of chronic inflammation in NAFLD and AATD. Activated liver macrophages promote inflammation characterized by cytokine and free radical (ROS) production and increased lipid oxidation. In this scenario, to diminish the net increment of lipids, hepatocytes fuse triglycerides (TAG) stored in lipid droplets into APOB-containing lipoproteins and increase the expression of the CD36 receptor to export the lipids out of the cells. In NAFLD patients, this increases the plasma lipoprotein levels with the concomitant risk of cardiovascular disease (CVD). In AATD patients, despite the increased expression of CD36, the accumulation of Z-AAT protein impairs lipids secretion and lowers the risk of CVD. LPL: lipoprotein lipase; HL: hepatic lipase; CETP: cholesteryl ester transfer protein; oxLDL: oxidized LDL.
4. Features of Lipid Metabolism in NAFLD and AATD
Lipids are key cellular components involved in maintaining the integrity of cellular membranes and energy homeostasis, although they also contribute to pathologies [63][41]. Lipid homeostasis in the liver depends on the equilibrated balance between lipid acquisition (de novo formation and uptake), storage, and removal [64][42]. Neutral lipids (sterol esters and triglycerides) are stored in LDs, and in a healthy liver, these lipids do not exceed 5% [65][43]. Fatty acids stored as sterol esters and triglycerides are used during liver homeostasis to generate energy via fatty acid oxidation or are transported to other organs in very-low-density lipoprotein (VLDL) [66,67][44][45] (Figure 1).
A composite route required for VLDL assembly is the lipidation of APOB100, a main and highly hydrophobic apolipoprotein. Initially, VLDLs are pre-assembled in ER lumen by the microsomal triglyceride transfer protein [68][46] and are subsequently moved to the secretory pathway as VLDL2 particles (TAG poor). These particles are secreted out of the hepatocytes or undergo additional lipidation through LD fusion and become VLDL1 particles (TAG enriched) [69][47].
NAFLD is a multifactorial disorder, in which genetic alterations play a role [72][48]. For example, genes such as PNPLA3 [73][49] and TM6SF2 [74][50] are linked to a high risk of NAFLD. The patatin-like phospholipase domain-containing 3 gene (PNPLA3) encodes a membrane-associated lipase that mediates triacylglycerol hydrolysis to manage the increasing amount of lipids after a meal intake. The nonsynonymous transversion from cytosine to guanine (rs738409) renders an amino acid change at codon 148 (isoleucine to methionine) that results in an imbalance between the liver triglyceride content and VLDL secretion [75][51].
5. Relationships between NAFLD, AATD, and Chronic Obstructive Pulmonary Disease (COPD)
NAFLD is a progressive liver disease evolving via NASH and fibrosis to cirrhosis, and eventually to hepatocellular carcinoma [89][52]. Investigations demonstrate that NAFLD, NASH, and liver fibrosis are prevalent in patients with COPD (by 41%, 37%, and 61%, respectively) [90][53]. Patients with COPD and NASH seem to have elevated TNF-α and leptin levels, unlike patients with COPD without liver damage [91][54]. It is thought that the chronic inflammatory synergy between NAFLD/NASH and COPD can trigger further injury and the progression of both diseases [91,92,93][54][55][56]. AATD is the most common genetic cause of emphysema, and, as a result, the lack of normal levels of AAT do not protect the lungs from damage, leading to an increased risk for developing COPD. Subjects with homozygous ZZ and heterozygous MZ AATD genotypes seem to also have a higher risk of developing NAFLD than non-deficient subjects [94][57]. For decades, intravenous therapy with human-plasma-purified AAT has been used to treat patients with AATD-related emphysema. The lung-protective effects of AAT are attributed to the inhibition of proteases involved in lung matrix fragmentation, macrophage activation, and endothelial cell apoptosis [95][58].6. Organoids to Model Liver Disease
In vitro two-dimensional (2D) cell models are widely used to reproduce the physiopathology and molecular mechanisms of various diseases. Traditionally used human cell lines are relatively cheap, easy to handle, and can be genetically modified. Nevertheless, the use of cell lines to address questions related to specific human diseases is not always straightforward, and they also lack tissue organization and grow without a physiological context. Therefore, the more recently developed three-dimensional (3D) cell cultures have become better experimental tools [103][59]. The 3D cultures known as organoids can be generated from adult stem cells, embryonic stem cells (ESCs), or induced pluripotent stem cells (iPSCs). Organoids derived from human progenitor cells can be long-term expanded; they can recapitulate organ architecture with remarkable fidelity, with the presence of multiple cell types of the specific organ [104,105][60][61]; and they assume at least some functions of the organ. Hepatic organoids generated from AATD patients have been proven as a new tool to study the pathophysiological characteristics of the liver [119,120][62][63]. These organoids have typical intrahepatic retention of Z-AAT polymers and show a positive diastase-resistant (PAS) staining [119,120][62][63]. Additionally, when AATD hepatic organoids were differentiated into hepatocytes, Huch and collaborators showed that these cells presented high ER stress and apoptosis [120][63]. Gomez-Mariano and colleagues further confirmed that differentiated liver organoids express albumin (ALB) and apolipoprotein B (APOB) genes, two specific hepatocyte markers [119][62]. Hepatocytes or hepatic parenchymal cells comprise approximately 60% of all liver cells, which form a 3D lattice filled by hepatic sinusoids. This latter provides nourishment for the parenchymal cells of the 3D structure, inter-luminal Kupffer, sinusoidal endothelial cells, and perisinusoidal stellate cells [121][64]. Different 3D cell strategies have been used for NAFLD modelling because the progression of this disease depends on the interactions among multiple liver cell types [122,123][65][66].7. Conclusions
NAFLD and AATD are hepatic diseases characterized by increased hepatic lipid content and consequently the intracellular accumulation of lipid droplets. To maintain liver homeostasis, hepatocytes in NAFLD eliminate the excess of lipids by exporting them to the bloodstream as lipoproteins, which results in an increased risk of cardiovascular disease in NAFLD patients. Conversely, in patients with AATD, the intrahepatic accumulation of misfolded AAT protein lowers lipid secretion and thus risk for cardiovascular disease. The use of patient-derived liver organoids as new cellular models is of great value, especially for the development of new personalized therapies, as well as for studying the underlying molecular mechanisms in NAFLD and AATD.References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatol. Baltim. Md. 2016, 64, 73–84.
- Teng, T.; Qiu, S.; Zhao, Y.; Zhao, S.; Sun, D.; Hou, L.; Li, Y.; Zhou, K.; Yu, X.; Yang, C.; et al. Pathogenesis and Therapeutic Strategies Related to Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 7841.
- Liebe, R.; Esposito, I.; Bock, H.H.; Vom Dahl, S.; Stindt, J.; Baumann, U.; Luedde, T.; Keitel, V. Diagnosis and Management of Secondary Causes of Steatohepatitis. J. Hepatol. 2021, 74, 1455–1471.
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A New Definition for Metabolic Dysfunction-Associated Fatty Liver Disease: An International Expert Consensus Statement. J. Hepatol. 2020, 73, 202–209.
- Eslam, M.; Sanyal, A.J.; George, J. International Consensus Panel MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1.
- Younossi, Z.M.; Rinella, M.E.; Sanyal, A.J.; Harrison, S.A.; Brunt, E.M.; Goodman, Z.; Cohen, D.E.; Loomba, R. From NAFLD to MAFLD: Implications of a Premature Change in Terminology. Hepatol. Baltim. Md. 2021, 73, 1194–1198.
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922.
- Hadizadeh, F.; Faghihimani, E.; Adibi, P. Nonalcoholic Fatty Liver Disease: Diagnostic Biomarkers. World J. Gastrointest. Pathophysiol. 2017, 8, 11–26.
- Targher, G.; Byrne, C.D.; Tilg, H. NAFLD and Increased Risk of Cardiovascular Disease: Clinical Associations, Pathophysiological Mechanisms and Pharmacological Implications. Gut 2020, 69, 1691–1705.
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Metabolism 2016, 65, 1038–1048.
- Harte, A.L.; da Silva, N.F.; Creely, S.J.; McGee, K.C.; Billyard, T.; Youssef-Elabd, E.M.; Tripathi, G.; Ashour, E.; Abdalla, M.S.; Sharada, H.M.; et al. Elevated Endotoxin Levels in Non-Alcoholic Fatty Liver Disease. J. Inflamm. 2010, 7, 15.
- Pendyala, S.; Walker, J.M.; Holt, P.R. A High-Fat Diet Is Associated with Endotoxemia That Originates from the Gut. Gastroenterology 2012, 142, 1100–1101.e2.
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.-L.; Gaudin, F.; Naveau, S.; Prévot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic Lipids Stored by Kupffer Cells Correlates with Their Pro-Inflammatory Phenotype at an Early Stage of Steatohepatitis. J. Hepatol. 2012, 57, 141–149.
- Walther, T.C.; Farese, R.V. Lipid Droplets And Cellular Lipid Metabolism. Annu. Rev. Biochem. 2012, 81, 687–714.
- Olzmann, J.A.; Carvalho, P. Dynamics and Functions of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155.
- Mashek, D.G. Hepatic Lipid Droplets: A Balancing Act between Energy Storage and Metabolic Dysfunction in NAFLD. Mol. Metab. 2021, 50, 101115.
- Yang, H.; Liu, J. Chapter 11-Structure and Function of Lipid Droplets. In Biochemistry of Lipids, Lipoproteins and Membranes, 7th ed.; Ridgway, N.D., McLeod, R.S., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 357–394. ISBN 978-0-12-824048-9.
- Janciauskiene, S.M.; Bals, R.; Koczulla, R.; Vogelmeier, C.; Köhnlein, T.; Welte, T. The Discovery of A1-Antitrypsin and Its Role in Health and Disease. Respir. Med. 2011, 105, 1129–1139.
- Seixas, S.; Marques, P.I. Known Mutations at the Cause of Alpha-1 Antitrypsin Deficiency an Updated Overview of SERPINA1 Variation Spectrum. Appl. Clin. Genet. 2021, 14, 173–194.
- Ogushi, F.; Fells, G.A.; Hubbard, R.C.; Straus, S.D.; Crystal, R.G. Z-Type Alpha 1-Antitrypsin Is Less Competent than M1-Type Alpha 1-Antitrypsin as an Inhibitor of Neutrophil Elastase. J. Clin. Investig. 1987, 80, 1366–1374.
- de Serres, F.J.; Blanco, I. Prevalence of A1-Antitrypsin Deficiency Alleles PI*S and PI*Z Worldwide and Effective Screening for Each of the Five Phenotypic Classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: A Comprehensive Review. Ther. Adv. Respir. Dis. 2012, 6, 277–295.
- Carlson, J.A.; Rogers, B.B.; Sifers, R.N.; Hawkins, H.K.; Finegold, M.J.; Woo, S.L. Multiple Tissues Express Alpha 1-Antitrypsin in Transgenic Mice and Man. J. Clin. Investig. 1988, 82, 26–36.
- Sinden, N.J.; Baker, M.J.; Smith, D.J.; Kreft, J.-U.; Dafforn, T.R.; Stockley, R.A. α-1-Antitrypsin Variants and the Proteinase/Antiproteinase Imbalance in Chronic Obstructive Pulmonary Disease. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2015, 308, L179–L190.
- Blanco, I.; Lipsker, D.; Lara, B.; Janciauskiene, S. Neutrophilic Panniculitis Associated with Alpha-1-Antitrypsin Deficiency: An Update. Br. J. Dermatol. 2016, 174, 753–762.
- Sun, R.; Xu, Z.; Zhu, C.; Chen, T.; Muñoz, L.E.; Dai, L.; Zhao, Y. Alpha-1 Antitrypsin in Autoimmune Diseases: Roles and Therapeutic Prospects. Int. Immunopharmacol. 2022, 110, 109001.
- Lomas, D.A.; Evans, D.L.; Finch, J.T.; Carrell, R.W. The Mechanism of Z Alpha 1-Antitrypsin Accumulation in the Liver. Nature 1992, 357, 605–607.
- Stoller, J.K.; Aboussouan, L.S. A Review of A1-Antitrypsin Deficiency. Am. J. Respir. Crit. Care Med. 2012, 185, 246–259.
- Hotamisligil, G.S. Inflammation and Metabolic Disorders. Nature 2006, 444, 860–867.
- Greenberg, A.S.; Obin, M.S. Obesity and the Role of Adipose Tissue in Inflammation and Metabolism. Am. J. Clin. Nutr. 2006, 83, 461S–465S.
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 Contributes to Macrophage Infiltration into Adipose Tissue, Insulin Resistance, and Hepatic Steatosis in Obesity. J. Clin. Investig. 2006, 116, 1494–1505.
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W. CCR2 Modulates Inflammatory and Metabolic Effects of High-Fat Feeding. J. Clin. Investig. 2006, 116, 115–124.
- Chakarov, S.; Lim, H.Y.; Tan, L.; Lim, S.Y.; See, P.; Lum, J.; Zhang, X.-M.; Foo, S.; Nakamizo, S.; Duan, K.; et al. Two Distinct Interstitial Macrophage Populations Coexist across Tissues in Specific Subtissular Niches. Science 2019, 363, eaau0964.
- Li, C.; Xu, M.M.; Wang, K.; Adler, A.J.; Vella, A.T.; Zhou, B. Macrophage Polarization and Metainflammation. Transl. Res. J. Lab. Clin. Med. 2018, 191, 29–44.
- Murray, P.J.; Wynn, T.A. Protective and Pathogenic Functions of Macrophage Subsets. Nat. Rev. Immunol. 2011, 11, 723–737.
- Odegaard, J.I.; Chawla, A. Alternative Macrophage Activation and Metabolism. Annu. Rev. Pathol. 2011, 6, 275–297.
- Pradere, J.-P.; Kluwe, J.; De Minicis, S.; Jiao, J.-J.; Gwak, G.-Y.; Dapito, D.H.; Jang, M.-K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic Macrophages but Not Dendritic Cells Contribute to Liver Fibrosis by Promoting the Survival of Activated Hepatic Stellate Cells in Mice. Hepatol. Baltim. Md. 2013, 58, 1461–1473.
- Hammoutene, A.; Rautou, P.-E. Role of Liver Sinusoidal Endothelial Cells in Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2019, 70, 1278–1291.
- Pigeolet, E.; Corbisier, P.; Houbion, A.; Lambert, D.; Michiels, C.; Raes, M.; Zachary, M.-D.; Remacle, J. Glutathione Peroxidase, Superoxide Dismutase, and Catalase Inactivation by Peroxides and Oxygen Derived Free Radicals. Mech. Ageing Dev. 1990, 51, 283–297.
- Clare, K.; Dillon, J.F.; Brennan, P.N. Reactive Oxygen Species and Oxidative Stress in the Pathogenesis of MAFLD. J. Clin. Transl. Hepatol. 2022, 10, 939–946.
- Song, M.J.; Malhi, H. The Unfolded Protein Response and Hepatic Lipid Metabolism in Non Alcoholic Fatty Liver Disease. Pharmacol. Ther. 2019, 203, 107401.
- Anand, P.K. Lipids, Inflammasomes, Metabolism, and Disease. Immunol. Rev. 2020, 297, 108–122.
- Krahmer, N.; Farese, R.V.; Walther, T.C. Balancing the Fat: Lipid Droplets and Human Disease. EMBO Mol. Med. 2013, 5, 905–915.
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD). European Association for the Study of Obesity (EASO) EASL-EASD-EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1388–1402.
- Tulenko, T. The Physiology of Lipoproteins. J. Nucl. Cardiol. 2002, 9, 638–649.
- Diffenderfer, M.R.; Schaefer, E.J. The Composition and Metabolism of Large and Small LDL. Curr. Opin. Lipidol. 2014, 25, 221–226.
- Olofsson, S.O.; Stillemark-Billton, P.; Asp, L. Intracellular Assembly of VLDL: Two Major Steps in Separate Cell Compartments. Trends Cardiovasc. Med. 2000, 10, 338–345.
- Stillemark-Billton, P.; Beck, C.; Borén, J.; Olofsson, S.-O. Relation of the Size and Intracellular Sorting of ApoB to the Formation of VLDL 1 and VLDL 2. J. Lipid Res. 2005, 46, 104–114.
- Anstee, Q.M.; Seth, D.; Day, C.P. Genetic Factors That Affect Risk of Alcoholic and Nonalcoholic Fatty Liver Disease. Gastroenterology 2016, 150, 1728–1744.e7.
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic Variation in PNPLA3 Confers Susceptibility to Nonalcoholic Fatty Liver Disease. Nat. Genet. 2008, 40, 1461–1465.
- Borén, J.; Adiels, M.; Björnson, E.; Matikainen, N.; Söderlund, S.; Rämö, J.; Ståhlman, M.; Ripatti, P.; Ripatti, S.; Palotie, A.; et al. Effects of TM6SF2 E167K on Hepatic Lipid and Very Low-Density Lipoprotein Metabolism in Humans. JCI Insight 2020, 5, e144079.
- Borén, J.; Adiels, M.; Björnson, E.; Matikainen, N.; Söderlund, S.; Rämö, J.; Henricsson, M.; Ripatti, P.; Ripatti, S.; Palotie, A.; et al. Effects of PNPLA3 I148M on Hepatic Lipid and Very-Low-Density Lipoprotein Metabolism in Humans. J. Intern. Med. 2022, 291, 218–223.
- Nati, M.; Haddad, D.; Birkenfeld, A.L.; Koch, C.A.; Chavakis, T.; Chatzigeorgiou, A. The Role of Immune Cells in Metabolism-Related Liver Inflammation and Development of Non-Alcoholic Steatohepatitis (NASH). Rev. Endocr. Metab. Disord. 2016, 17, 29–39.
- Moon, S.W.; Kim, S.Y.; Jung, J.Y.; Kang, Y.A.; Park, M.S.; Kim, Y.S.; Chang, J.; Ro, J.S.; Lee, Y.-H.; Lee, S.H. Relationship between Obstructive Lung Disease and Non-Alcoholic Fatty Liver Disease in the Korean Population: Korea National Health and Nutrition Examination Survey, 2007–2010. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 2603–2611.
- Viglino, D.; Plazanet, A.; Bailly, S.; Benmerad, M.; Jullian-Desayes, I.; Tamisier, R.; Leroy, V.; Zarski, J.-P.; Maignan, M.; Joyeux-Faure, M.; et al. Impact of Non-Alcoholic Fatty Liver Disease on Long-Term Cardiovascular Events and Death in Chronic Obstructive Pulmonary Disease. Sci. Rep. 2018, 8, 16559.
- Viglino, D.; Jullian-Desayes, I.; Minoves, M.; Aron-Wisnewsky, J.; Leroy, V.; Zarski, J.-P.; Tamisier, R.; Joyeux-Faure, M.; Pépin, J.-L. Nonalcoholic Fatty Liver Disease in Chronic Obstructive Pulmonary Disease. Eur. Respir. J. 2017, 49, 1601923.
- Rodríguez-Roisin, R.; Krowka, M.J. Hepatopulmonary Syndrome--a Liver-Induced Lung Vascular Disorder. N. Engl. J. Med. 2008, 358, 2378–2387.
- Cheeney, G.; Pac, L.J.; Gopal, P.; Landis, C.S.; Konnick, E.Q.; Swanson, P.E.; Greene, D.N.; Lockwood, C.M.; Westerhoff, M. Increased Frequency of Heterozygous Alpha-1-Antitrypsin Deficiency in Liver Explants From Nonalcoholic Steatohepatitis Patients. Liver Transpl. 2020, 26, 17–24.
- Lockett, A.D.; Kimani, S.; Ddungu, G.; Wrenger, S.; Tuder, R.M.; Janciauskiene, S.M.; Petrache, I. A₁-Antitrypsin Modulates Lung Endothelial Cell Inflammatory Responses to TNF-α. Am. J. Respir. Cell Mol. Biol. 2013, 49, 143–150.
- Schutgens, F.; Clevers, H. Human Organoids: Tools for Understanding Biology and Treating Diseases. Annu. Rev. Pathol. 2020, 15, 211–234.
- Lancaster, M.A.; Huch, M. Disease Modelling in Human Organoids. Dis. Model. Mech. 2019, 12, dmm039347.
- Marsee, A.; Roos, F.J.M.; Verstegen, M.M.A.; Gehart, H.; de Koning, E.; Lemaigre, F.; Forbes, S.J.; Peng, W.C.; Huch, M.; Takebe, T.; et al. Building Consensus on Definition and Nomenclature of Hepatic, Pancreatic, and Biliary Organoids. Cell Stem Cell 2021, 28, 816–832.
- Gómez-Mariano, G.; Matamala, N.; Martínez, S.; Justo, I.; Marcacuzco, A.; Jimenez, C.; Monzón, S.; Cuesta, I.; Garfia, C.; Martínez, M.T.; et al. Liver Organoids Reproduce Alpha-1 Antitrypsin Deficiency-Related Liver Disease. Hepatol. Int. 2020, 14, 127–137.
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.A.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-Term Culture of Genome-Stable Bipotent Stem Cells from Adult Human Liver. Cell 2015, 160, 299–312.
- Trefts, E.; Gannon, M.; Wasserman, D.H. The Liver. Curr. Biol. 2017, 27, R1147–R1151.
- Wang, Y.; Wang, H.; Deng, P.; Tao, T.; Liu, H.; Wu, S.; Chen, W.; Qin, J. Modeling Human Nonalcoholic Fatty Liver Disease (NAFLD) with an Organoids-on-a-Chip System. ACS Biomater. Sci. Eng. 2020, 6, 5734–5743.
- Carter, J.K.; Friedman, S.L. Hepatic Stellate Cell-Immune Interactions in NASH. Front. Endocrinol. 2022, 13, 867940.
More