Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Kenneth K. Wu and Version 2 by Sirius Huang.
Extracellular succinate induces cellular changes and tissue adaptation or damage by ligating cell surface succinate receptor-1 (SUCNR-1) and activating downstream signaling pathways and transcriptional programs.
- extracellular succinate
- succinate receptor-1
- succinate dehydrogenase
- inflammation
- fibrosis
- myocardial infarction
1. Introduction
Succinic acid is a tricarboxylic acid (TCA) cycle metabolite that is derived from succinyl CoA and oxidized to form fumarate [1]. It is normally confined to the mitochondrial matrix and its level is tightly regulated [1]. When cells are stressed, disruption of the TCA cycle may result in elevation of succinate in the matrix and the leakage of excessive succinate into the cytoplasm, where it acts as a signaling molecule to impact diverse cellular functions through inhibition of a large group of 2-oxoglutarate-dependent dioxygenases (2OGDD), notably prolyl hydroxylase (PHD) and ten–eleven translocation (TET) [2][3][2,3]. Inhibition of PHD by cytosolic succinate leads to impaired degradation of hypoxia inducible factor-1 (HIF-1α) and HIF-1α-mediated changes in transcription of metabolic enzymes, angiogenic factors, and pro-inflammatory mediators [4][5][6][7][4,5,6,7]. Inhibition of TET-2, on the other hand, results in impaired hydroxylation of DNA methyl groups, and, consequently, DNA hypermethylation, which is associated with tumorigenesis and cancer metastasis [8][9][8,9]. Cytosolic succinate is secreted into the extracellular space and diffused into the circulating blood, where it acts as a local and/or systemic autacoid, regulating physiological functions and pathological processes. Cytosolic succinate has been covered in excellent review articles [10][11][10,11].
2. Skeletal Muscle-Derived Extracellular Succinate Confers Physiological Adaptation to Exercise and Cold Exposure
Extracellular succinate is a crucial messenger to facilitate adaptation to physiological stresses, such as cold exposure, vigorous exercise, and physical activity. Skeletal muscle plays a central role in providing extracellular succinate to drive the adaptation.
2.1. Extracellular Succinate Promotes Muscle Remodeling
It was reported almost a half century ago that aerobic exercise increased blood levels of succinate [12]. Subsequent studies confirmed the accumulation of succinate during exercise. A meta-analysis of relevant exercise-associated metabolomic studies reveals that exercise increase circulating metabolites, including succinate [13]. A recent report indicates that, in response to exercise, succinate is secreted from skeletal muscle cells via monocarboxylate transporter-1 (MCT-1), and the secreted succinate activates non-myofibrillar resident cells such as stromal cells, endothelial cells, and satellite cells in skeletal muscle to promote skeletal muscle remodeling and innervation via succinate receptor-1 (SUCNR-1) [14]. It is unclear how exercise induces succinate accumulation. One possible explanation is that exercise induces muscle cell metabolic reprogramming, leading to increased glycolysis and a disrupted TCA cycle, such as inhibition of succinate dehydrogenase (SDH). SDH possesses dual activities: it catalyzes oxidation of succinate to form fumarate in the TCA cycle and functions as complex II in the ETC to convert ubiquinone to ubiquinol for oxidative phosphorylation and ATP generation [15][16][15,16]. Akin to exercise-induced skeletal muscle metabolic changes, LPS-stimulated macrophages exhibit a metabolic shift to aerobic glycolysis and perturbation of the TCA cycle characterized by blocking isocitrate dehydrogenase activity, which leads to itaconate accumulation [17]. Excessive itaconate inhibits SDH catalytic activity, resulting in succinate accumulation [17]. Contrary to this possible mechanism is a report that congenital deficiency of SDH in a patient rendered intolerance to exercise due to early skeletal muscle fatigue and weakness during exercise [18][19][18,19]. It was proposed that TCA enzyme deficiency impairs oxidative phosphorylation and reduces energy supply. Succinate supplementation was reported to enhance muscle fiber oxidative phosphorylation and increase muscle strength and endurance [20][21][20,21]. The reason for the different roles that extracellular succinate plays in muscle strength and endurance during exercise is unclear. Further studies are needed to elucidate the underlying mechanism.
2.2. Extracellular Succinate Upregulates Adipose Tissue Energy Expenditure and Thermogenesis
Brown adipose tissues (BATs) act as a central regulator of energy expenditure for thermogenesis and play an important role in maintaining body temperature during cold exposure [22][23][22,23]. Metabolomic analysis identifies succinate as a key driver of energy expenditure in BAT [24]. It has been reported that cold-exposure-associated muscular shivering leads to release of succinate from muscle fibers, resulting in elevation of succinate levels in the circulating blood [24]. The extracellular succinate is taken up by brown adipocytes and enters the mitochondria, where it replenishes the substrate for SDH and complex II to generate ROS and stimulate uncoupled respiration in an uncoupled protein-1 (UCP-1)-dependent manner [24]. Thermogenesis from the UCP-1 pathway is abrogated by either pharmacological inhibition of muscle contraction or SDH/complex II. It is to be noted that BAT UCP-1 is a key player in controlling the level of succinate in circulating blood. Genetic deletion of UCP-1 in mice results in the elevation of blood succinate levels, which contributes to liver inflammation and fibrosis [25].
In summary, skeletal muscle cells are in a pivotal position to supply extracellular succinate, which acts as an intercellular messenger to enhance muscle endurance during vigorous exercise and generate heat during cold exposure (Figure 1).
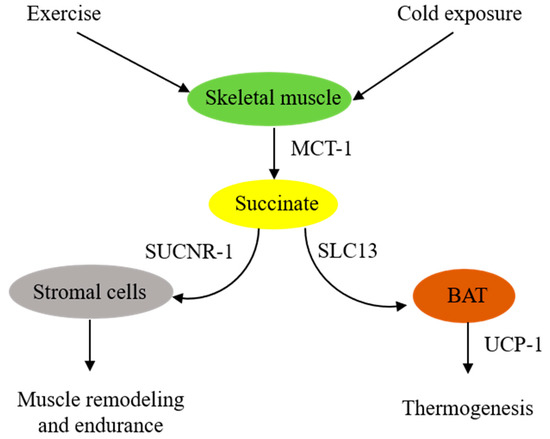
Figure 1. SchematicSchematic illustration of extracellular succinate as a physiological messenger. illustration of extracellular succinate as a physiological messenger. Physiological stresses such as vigorous exercise or cold exposure induce skeletal muscle fibers to secrete succinate, which acts as a messenger to confer physiological adaptation.
3. Extracellular Succinate Mediates Diverse Pathophysiological Processes via SUCNR-1
As described above, extracellular succinate serves as a substrate supplement via anaplerosis to confer thermogenesis for adaptation to cold and muscle remodeling to enhance endurance during vigorous exercise. However, extracellular succinate mediates diverse pathophysiological processes and exacerbates human diseases through interaction with a membrane G-protein-coupled receptor (GPCR), GPR91. In searching for natural ligands for orphan GPCRs, He et al. identified succinate from animal kidney extracts as a selective ligand for GPR91, an orphan GPCR with sequence homology to purinergic receptors [26][27][28][26,27,28]. GPR91 was shown to be expressed in the juxtaglomerular apparatus, especially the afferent arterial endothelial cells, and involved in inducing renin production and secretion [29][30][29,30]. It was linked to diabetes-associated hypertension, as elevated succinate in diabetes stimulates renin production via GPR91 [29][30][29,30]. GPR91 was detected in proximal and distal duct epithelial cells in kidneys and was considered to play a role in regulating renal tubular functions, although the exact activity remains to be ascertained [31]. Further studies reveal that GPR91 expression is not restricted to renal tissues. In fact, it is widely distributed on different cells where it carries out tissue-specific functions and mediates pathophysiological processes in human diseases. As GPR91 proves to be a selective succinate receptor, it is commonly called SUCNR-1. Succinate-ligated SUCNR-1 is signaled via Gi and Gq with the activation of cardinal signaling pathways, resulting in elevation of intracellular calcium and inositol triphosphate (IP3), reduction of cyclic AMP, and activation of ERK1/2 [27]. Under certain circumstances, succinate-activated SUCNR-1 may signal via Gq, as reported in macrophages by [32]. SUCNR-1 mediates physiological roles, such as immune responses, glucose homeostasis, hematopoiesis, and platelet aggregation, and pathological conditions, such as tissue injury, inflammation, fibrosis, and cancer metastasis [33][34][35][36][37][38][33,34,35,36,37,38]. The succinate-SUCNR-1 axis plays a critical role in important human diseases (Figure 2).
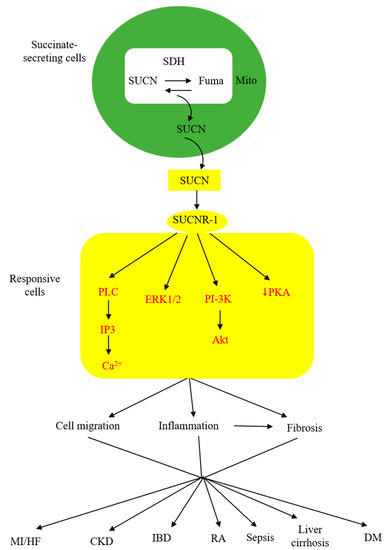
Figure 2. Schematic illustration of the involvement of succinate (SUCN)-SUCNR-1 axis in diverse pathological processes and human diseases. MI, myocardial infarction; HF, heart failure; CKD, chronic kidney disease; IBD, inflammatory bowel disease; DM, diabetes mellitus.
4. Succinate Elicits Intestinal Immunity and Triggers Colon Inflammation
Under normal conditions, the intestinal lumen and feces contain low levels of succinate, despite a large quantity of succinate produced by microbial fermentation of dietary fibers [39]. Low luminal succinate is attributed to the conversion of succinate to short-chain fatty acids (acetate, propionate, and butyrate) by succinate-consuming bacteria in the gut microbiota [40][41][40,41]. Intestinal epithelial cells express solute carrier 13A (SLC13A) family proteins, notably SLC13A-2, -3, and -5, which take up luminal succinate [42]. It was reported that small intestinal epithelial cells take up succinate and convert it to glucose via gluconeogenesis [43], which exerts a great impact on reducing lipid deposition and attenuating hepatic inflammation [44]. Despite a low level of succinate in the lumen of the small intestine, succinate derived from the diet or protist and helminth infestation interacts with SUCNR-1 expressed on the tuft cells and transmits signals to secrete IL-25, which induces tuft cell and goblet cell hypertrophy and activates type 2 innate lymphoid cells (ILC2) to elicit type II immunity [45][46][47][45,46,47].
By contrast, succinate accumulation in the colon exerts a detrimental effect on the epithelium. Perturbation of colon microbiota by antibiotics, high-fat diet, or inflammatory mediators results in increasing succinate-producing and diminishing succinate-consuming strains in the colon microbiome and the consequent overproduction of succinate [48][49][48,49]. Unlike the small intestine, which contains succinate-sensing tuft cells, the colon epithelium contains few, if any, tuft cells and does not elicit immunity. The accumulated luminal succinate disrupts epithelial barrier function and induces inflammation and fibrosis by activating macrophages and fibroblasts, which reside in the subepithelial region [49]. Chronic succinate elevation in the colon lumen was reported to induce colitis in mice [49]. Succinate causes epithelial damage, macrophage activation, and fibroblast transdifferentiation by ligating SUCNR-1 and activating SUCNR-1-mediated signaling pathways. One of the mechanisms by which luminal succinate reaches the subepithelial space is through a transepithelial pathway in which succinate is taken up by epithelial cells via Slc13A transporters, such as Slc13A-2 or A-3, and secreted into the extracellular space via the organic ion transporters [42]. Extracellular succinate is taken up by the infiltrating macrophages, and elevated cytosolic succinate enhances inflammation through inhibition of PHD, thereby stabilizing HIF-1α, which mediates IL-1β release [7][50][7,50]. As illustrated in Figure 3, microbiota-derived succinate is handled differently in the small intestine vs. the colon, carries out physiological functions in the small intestine via tuft cells, and induces colon epithelial cell damage and subepithelial inflammation and fibrosis by activating macrophages and fibroblasts.
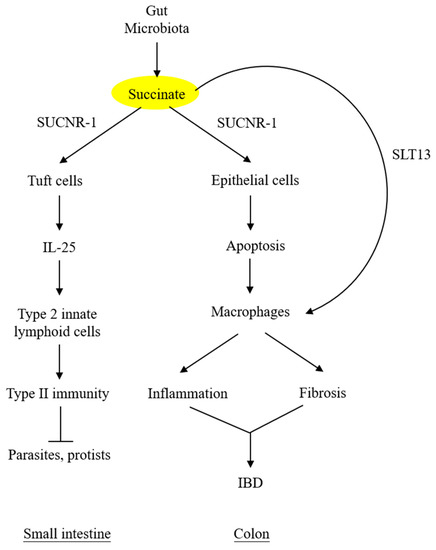
Figure 3. DifferentDifferent roles of microbiota-derived succinate in small intestine vs. colon. roles of microbiota-derived succinate in small intestine vs. colon. Succinate accumulation in colon exerts a direct detrimental effect on colon epithelium. It may also activate subepithelial macrophages via transepithelial transport. SLT13, solute transport 13; IBD, inflammatory bowel disease.
Serum and fecal succinate levels are elevated in humans with inflammatory bowel diseases, i.e., Crohn’s disease and ulcerative colitis [42][49][42,49]. SUCNR-1 is detected on epithelial cells as well as inflammatory cells and fibroblasts in the lamina propria of IBD colonic tissues [42][49][42,49]. Importantly, SUCNR-1 expression is correlated with fibrosis [49]. Taken together, microbiota-derived succinate plays an important role in aggravating inflammation and inducing fibrosis in human IBD via SUCNR-1.