Intracellular chloride levels in the brain are regulated primarily through the opposing effects of two cation-chloride co-transporters (CCCs), namely K+-Cl− co-transporter-2 (KCC2) and Na+-K+-Cl− co-transporter-1 (NKCC1). These CCCs are differentially expressed throughout the course of development, thereby determining the excitatory-to-inhibitory γ-aminobutyric acid (GABA) switch. GABAergic excitation (depolarisation) is important in controlling the healthy development of the nervous system; as the brain matures, GABAergic inhibition (hyperpolarisation) prevails. This developmental switch in excitability is important, as uncontrolled regulation of neuronal excitability can have implications for health.
- GABAergic
- Na+-K+-2Cl− cotransporter 1 (NKCC1)
- K+-2Cl− cotransporter 2 (KCC2)
- Chloride (Cl−) homeostasis
- Huntington’s disease
- sleep disorders
The following contents are excerpts from your papers. They are editable. The entry will better spread your research and findings. We'll help to correct the format after your submission.
1. Introduction
GABA signalling is crucial in both motor and behavioural control; GABAergic neurotransmission is altered in HD [1][2][2,29]. To better understand GABAergic activity, we need to consider both K+-2Cl− cotransporter 2 (KCC2) expression and the maintenance of neuronal intracellular chloride (Cl−) concentration ([Cl−]i). Cl− is an important anion involved in the regulation of cell volume[3] [30], proliferation, and apoptosis[4] [31]. Cl− has a further role in determining membrane potential and the firing of action potentials[5] [32]. Extracellular [Cl−] tends to be fixed, while [Cl−]i is more variable[6] [33]. The presence of chloride-cation cotransporters (CCCs) is central to determining [Cl−]i[7] [34]. Such transporters are responsible for the bidirectional movement of Cl−, and its function is determined by the direction of flux[4] [31].
GABA is the main inhibitory neurotransmitter in the brain[8] [35]. GABA binds GABA type A receptors (GABAAR); these receptors are ligand-gated anion channels central to the control of Cl− movement[8] [35]. Noteworthily, GABAA receptors are permeable for both Cl− and bicarbonate (HCO3−); the net effect of GABA therefore also depends on the distribution of bicarbonate[9] [36]. Previous studies have also implicated HCO3− in GABAA receptor-mediated depolarization[10][11][12][13][14] [37,38,39,40,41]. In a recent study, Lombardi et al.[14] [41] suggest that implementation of physiological levels of HCO3−-conductivity to GABAA receptors enhances the [Cl−]i changes over an extensive range of [Cl−]I; however, this outcome strictly depends on the stability of the HCO3− gradient and the intracellular pH. For in-depth understanding on the relationship between distribution of HCO3− and GABA signalling, readers are referred to recent review on the subject[9][15][16][17] [36,42,43,44]. Yet, the reversal potential (when net flow = 0) for GABA (EGABA) is primarily determined by the reversal potential for Cl−; GABAergic signalling is therefore dependent on [Cl−]i[18] [45]. Whilst inhibitory GABAergic activity is important for proper central nervous system (CNS) functioning[19] [46], GABA can also induce membrane depolarisation[20] [47]. The excitatory action of GABA is important in the development of the nervous system [6][35]; its roles include regulating synaptogenesis in addition to supporting neurite outgrowth and the maturation of the neuronal network[1][6] [2,35]. High [Cl−]i produces less negative GABA currents that culminate in depolarisation events (excitation)[11] [48]. Conversely, low [Cl−]i leads to hyperpolarisation as a result of more negative EGABA values (inhibition) [18][45]. During development there is a gradual hyperpolarising shift in EGABA as a result of decreased [Cl−]i, which is maintained in the mature mammalian brain[22][23] [49,50]. Since the healthy brain relies on the proper balance between excitatory and inhibitory inputs, uncontrolled regulation of neuronal excitability can have implications for health[3] [32].
Neuronal [Cl−]i is largely regulated through the activity of Na+-K+-2Cl− cotransporter 1 (NKCC1) and KCC2 [24][25][26][51,52,53]. NKCC1 pumps Cl− into neurons, while KCC2 is responsible for Cl− efflux[23][26] [50,53]. Moreover, the extensively studied CCC family member, NKCC1, has numerous physiological obligations[27][28][29] [54,55,56] that make it a promising neurological drug target, owing to its importance in GABAergic signalling[23] [50]. Recently, Chew and colleagues[30] [57] determined the cryo-electron microscopy structure of NKCC1 from Danio rerio. This extensive study revealed the mechanisms involved in NKCC1 molecular transportation and communication and further provided insights into ion selectivity as well as coupling and translocation; a clearer framework for understanding the physiological functions of NKCC1 in relation to human diseases was also established[30] [57]. Besides, modulation of NKCC1 activity alongside that of KCC2 has been implicated in the development and progression of HD[1][31][32][33] [2,58,59,60]. These CCCs are differentially expressed over the course of development, and so the activity of KCC2 and NKCC1 is not synonymous between immature and mature neurons[23] [50]. In the embryonic and early postnatal period, solute carrier family 12 (SLC12), member A1 (SLC12A1) messenger RNA (mRNA) expression of NKCC1 is high[23] [50]. As maturation proceeds, NKCC1 expression decreases and SLC12, member A5 (SLC12A5) expression of mRNA encoding KCC2 is upregulated: there is a resultant net decrease in [Cl−]i[23] [50]. The developmental stimulation of KCC2 and inhibition of NKCC1 expression initiates the switch from excitatory to inhibitory GABA signalling [24][61]. These evolutionarily conserved transporters (KCC2 and NKCC1) are inclusive among central mediators of ion transport in multicellular organisms, with specific roles in regulating ionic and water homeostasis in mammalian CNS[25] [62], which is essential in determining the polarity of the neurons[26] [63]. Notably, during development, [Cl−]i increment is prominent in immature neurons and when activated, they display a depolarising response, which is due to the elevated expression of NKCC1 in comparison with KCC2[23] [50]. However, during maturation, NKCC1 expression gradually decreases, and KCC2 expression increases, resulting in an opposite expression pattern[23][36] [50,63] (also see Figure 2). Importantly, the inhibition/stimulation of KCC2/NKCC1 pair via protein phosphorylation is through a regulatory mechanism that works in a reciprocal pattern[26][37] [53,64] and members of the with-no-lysine kinase (WNKs) family as well as their downstream targets; STE20/SPS1-related proline/alanine rich kinase (SPAK) and oxidative stress response kinase (OSR1) are the most prominent kinases that regulate this process[25][37 [52,64,65,66,67][38][39][40]. Consequently, impaired ion homeostasis resulting from mutation in the physiological function of some of this transporter pair and/or their upstream regulators may be detrimental and subsequently result in diminished inhibition and augmented network hyperexcitability, which underlies numerous neurological disorders[25][39][41][42][43][44] [52,66,68,69,70,71] including HD[31][32][33] [58,59,60].
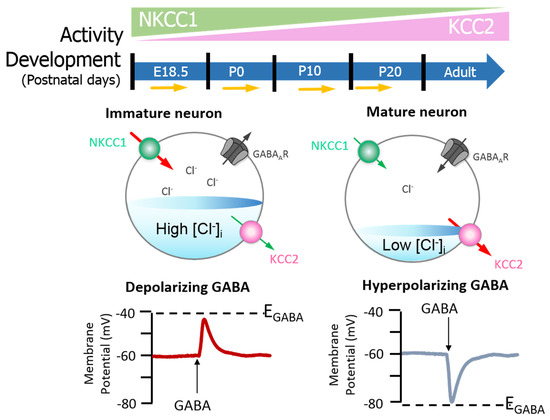
Figure 2. GABAA signalling shifts from depolarizing to hyperpolarising responses are mediated by developmental expression of KCC2 and NKCC1 in the brain (neocortical neurons) of rats. The differential expression of these channels regulates intracellular Cl− concentration ([Cl−]i) and therefore determines the activity of γ-aminobutyric acid (GABA). Na+–K+–Cl− cotransporter 1 (NKCC1) pumps Cl− into neurons; its expression is high in the early postnatal period, decreasing as maturation proceeds. The expression pattern for K+–2Cl− cotransporter 2 (KCC2), responsible for Cl− efflux, is directly opposite. In the embryonic and early postnatal periods, [Cl−]i is high, and so GABAergic signalling is excitatory (depolarising); as maturation occurs, [Cl−]i decreases, initiating the development hyperpolarising shift, whereby GABAergic signalling becomes inhibitory. Figure elements were taken and modified from Tillman and Zhang[26] [63].
Indeed, loss of KCC2 has implications in disease: KCC2 dysfunction and/or deficiency attenuates Cl− efflux and GABAergic inhibition is therefore impaired[5][19] [32,46]. When [Cl−]i exceeds equilibrium, depolarisation events contribute to the onset of neurological disease[8][19] [35,46]. Decreased KCC2 expression coupled with increased NKCC1 expression and/or activity has been documented in several pathologies[36][41 [63,68,69,71][42][44], including HD[31][32][33] [58,59,60]. In HD, HTT is mutated (mHTT) and acts to alter KCC2 and NKCC1 expressions and activity[31][32] [58,59] through mechanisms that remain undetermined. Since KCC2 and NKCC1 expressions and functionality are crucial in determining the effects of GABA dysregulated KCC2 and NKCC1 activities[3][19][45] [32,46,58,72], subsequent abnormalities in GABAergic signalling are thought to contribute to HD pathogenesis[1] [2]. Hence, this review aims to investigate the possible mechanisms by which KCC2 expression and function is altered in HD. Although HD is primarily characterised by uncoordinated motor activity [9,10], patients have additional, co-existing neurological disorders [17,18,19,20,21]. In view of the aforesaid, the association between altered KCC2 activity, and the comorbidities that present as part of the disease process will also be discussed. For examples, how are NKCC1 and KCC2 expression and activity controlled in the healthy brain? What are the known mechanisms by which KCC2 expression and activity are altered in HD? Additionally, how does altered KCC2 expression and activity contribute to HD comorbidities with a particular focus on cognitive and sleep changes?
2. KCC2 Regulation and Function in the Healthy Brain
Phosphorylation Regulation of KCC2 by Protein Kinase Signalling Pathways
The WNK-SPAK/OSR1 phosphorylate threonine residues 906 and 1007 (T906/T1007) and subsequently downregulate KCC2 mRNA gene expression; thus, a decline in its physiological function is observed[46][47] [73,74]. The phosphorylation of these residues is highest in the early postnatal period, with a gradual decrease throughout development[46][47] [73,74]; WNK1 activity is at its most reduced level in mature neurons[37][47] [64,74]. KCC2-T906/T1007 phosphorylation has been shown to decrease by approximately 95% between embryonic day 18.5 (E18.5) and adulthood in mice[48] [75]. This decrease in threonine phosphorylation may contribute to the developmental onset of KCC2 function[46][47] [73,74], thereby facilitating the upregulation of Cl− extrusion from mature neurons resulting in the hyperpolarising EGABA shift[5][49] [32,76] (also see Figure 3).
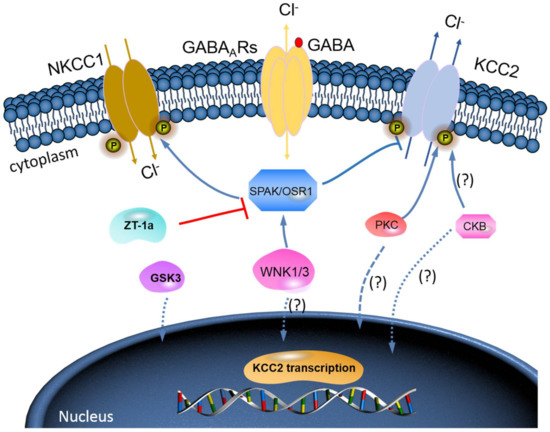
Figure 3. A novel strategy to facilitate neuronal Cl− extrusion and EGABA by coincident NKCC1 inhibition and KCC2 activation by inhibiting the WNK-SPAK/OSR1 kinases. Mammalian neurons that are challenged with multiple neuropsychiatric conditions (such as seizures, neuropathic pain, spasticity, schizophrenia, and others) are usually driven by hyperexcitable circuits, intraneuronal Cl− ([Cl−]i) levels are elevated due to increased NKCC1 activity, and/or decreased KCC2 activity, promoting GABAAR-mediated membrane depolarization and excitation. In healthy mature neurons, [Cl−]i is low due to the opposite activity profile of the CCCs, promoting GABAAR-mediated hyperpolarization, which is critical for the proper balance of excitation–inhibition in neuronal circuits. WNK-SPAK/OSR1 inhibition, via the coincident effects of NKCC1 inhibition and KCC2 activation (the main Cl− extrusion mechanism in neurons), might be a potent way of facilitating neuronal Cl− extrusion to restore ionic inhibition in diseases that are characterized by disordered Cl− homeostasis and GABA disinhibition. ZT-1a, a novel molecular compound, can specifically inhibit SPAK signalling pathway, thus interfering SPAK regulation of GABA signalling via NKCC1 inhibition and KCC2 activation[59] [86]. Activation of protein kinase C (PKC) and brain-type creatine kinase (CKB) are likely to increase KCC2 cell surface expression, but the mechanisms involved are still unclear.
Moore et al. [49] [76] demonstrated that preventing KCC2-T906/T1007 phosphorylation in vivo (assessed in knock-in mice), via threonine to alanine mutation, accelerates the onset of KCC2 function in the postnatal period. In the study, EGABA values were found to be hyperpolarised across neuronal development (patch-clamp experiment; cultured hippocampal knock-in mouse neurons), i.e., in preventing the phosphorylation of KCC2-T906/T1007, postnatal GABAergic depolarisation activity was largely abolished, thus suggesting that the developmental onset of hyperpolarising synaptic inhibition is dependent on regulated KCC2 phosphorylation[49] [76], and this is further supported by other studies[46][47] [73,74]. Therefore, potentiating KCC2 function to rescue delayed EGABA shift during development may improve cognitive defects[49] [76]. Serine 940 (S940) is another key phosphorylation site in the regulation of KCC2 activity, phosphorylation of S940 is controlled by protein kinase C (PKC)[50][51] [77,78]. S940 phosphorylation leads to decreased KCC2 internalisation and subsequent increased Cl− extrusion[34] [61], thereby increasing KCC2 function [34][50][61,77]. In this regard, Moore et al.[49] [76] further suggest that S940 can be experimentally mutated to alanine (S940A) to prevent its phosphorylation to facilitate [Cl−] increment. Briefly, they demonstrated that developmental EGABA shift is delayed in S940A neurons compared to wildtype (WT) controls, thereby suggesting that phospho-regulation of KCC2-S940 may be involved in defining the developmental onset of GABAergic inhibition[49] [76].
Furthermore, brain-type creatine kinase (CKB) also plays a vital role in regulating cellular energy homeostasis via ATP-dependent phosphorylated catalysis of creatine into phosphocreatine, thereby establishing a readily available ATP-buffering system[52] [79]. Notably, ATP is involved in the activation of Na+-K+-ATPase, which serves as a driving force for KCC activation; hence, it is expected that ATP should enhance the function of KCC2[53] [80]. Interestingly, some reports have affirmed an ATP-induced KCC activation [27,28]. Aside from potentially providing ATP, Hemmer et al.[54] [81] hypothesize that CKB might phosphorylate KCC2 to change its function, because CKB possesses autophosphorylation activity. However, the implication of the interaction between KCC2 and CKB in relation to their physiological functions and how intracellular ATP concentrations might contribute to KCC2 function is still elusive [53][80]. More importantly, however, the fact that WNK-SPAK/OSR1 kinase complex is known to phosphorylate and inhibit KCC2 or stimulate NKCC1[25][37][38][39][40] [52,64,65,66,67] is already established. Thus, molecular compounds that can block WNK-SPAK/OSR1 signalling pathway will result in activating KCC2 and inhibiting NKCC1 activities. The manipulation of the interaction between CKB and KCC2 activities could be a substitute mechanism to achieve KCC2 activation [53][55][80,82]. In fact, the interaction between CKB and KCC2 expression/activity has been implicated in the modulation of GABAAR-mediated signalling[1][32] [2,59]. Furthermore, previous reports have demonstrated that enhancement of CKB activity may facilitate the activation of KCC2 function[55][56][57][58] [82,83,84,85]. In HD, reduced expression and activity of CKB is associated with motor deficits and hearing impairment[56][57] [83,84]. By and large, the enhancement of CKB activity prior to its interaction with KCC2 activates its function resulting from inhibited phosphorylation of the WNK-SPAK/OSR1 signalling pathway may be a hypothesis worthy of intensive investigations (Figure 3). Hence, it is worthwhile to further investigate the interaction of KCC2 and CKB and how the interaction can modulate the WNK-SPAK/OSR1 signalling cascades in neurological diseases including HD.
Indeed, phosphorylation status of key regulatory sites on KCC2 determines when the developmental EGABA shift occurs, and regulated depolarising GABAergic signalling (largely in the early postnatal period) is necessary for normal cognitive and behavioural development[49] [76]. Since the phosphorylation process is central to KCC2 function, future research should assess whether the phosphorylation status of key KCC2 sites is constant between HD patients and controls. Further to this, it should be established if phosphorylation status changes as HD progresses. If the phosphorylation of key KCC2 sites does not occur as normal in HD gene carriers and patients, investigating how this affects the development hyperpolarising shift in EGABA is of concern; this research may provide an explanation for the behavioural and cognitive manifestations observed in HD patients. Investigation into the phosphorylation of KCC2 and the EGABA is of increasing interest, especially since it has been suggested that the potentiation of KCC2 function (accelerating hyperpolarising shift) can improve cognitive decline[49] [76].
Reference (we'll rearrange the references after you submitted it)
- Payne, J.A. Functional characterization of the neuronal-specific K-Cl cotransporter: Implications for [K+] oregulation. Am. J. Physiol. Cell Physiol. 1997, 273, C1516–C1525.
- Hsu, Y.T.; Chang, Y.G.; Chern, Y. Insights into GABAAergic system alteration in Huntington′s disease. Open Biol. 2018, 8.
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington′s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb. Perspect. Med. 2017, 7.
- Rosenblatt, A.; Brinkman, R.R.; Liang, K.Y.; Almqvist, E.W.; Margolis, R.L.; Huang, C.Y.; Sherr, M.; Franz, M.L.; Abbott, M.H.; Hayden, M.R.; et al. Familial influence on age of onset among siblings with Huntington disease. Am. J. Med. Genet. 2001, 105, 399–403.
- Genetic Modifiers of Huntington’s Disease Consortium. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell 2015, 162, 516–526.
- Moss, D.J.H.; Pardiñas, A.F.; Langbehn, D.; Lo, K.; Leavitt, B.R.; Roos, R.; Durr, A.; Mead, S.; Holmans, P.; Jones, L.; et al. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. 2017, 16, 701–711.
- Georgiou, N.; Bradshaw, J.L.; Chiu, E.; Tudor, A.; O′Gorman, L.; Phillips, J.G. Differential clinical and motor control function in a pair of monozygotic twins with Huntington’s disease. Mov. Disord. 1999, 14, 320–325.
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228.
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981.
- Goodman, A.O.; Rogers, L.; Pilsworth, S.; McAllister, C.J.; Shneerson, J.M.; Morton, A.J.; Barker, R.A. Asymptomatic sleep abnormalities are a common early feature in patients with Huntington’s disease. Curr. Neurol. Neurosci. Rep. 2011, 11, 211–217.
- Pellegrino, C.M.; Rybicki, A.C.; Musto, S.; Nagel, R.L.; Schwartz, R.S. Molecular identification and expression of erythroid K: Cl cotransporter in human and mouse erythroleukemic cells. Blood Cells Mol. Dis. 1998, 24, 31–40.
- Russell, J.M. Sodium-potassium-chloride cotransport. Physiol. Rev. 2000, 80, 211–276.
- Schulte, J.T.; Wierenga, C.J.; Bruining, H. Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci. Biobehav. Rev. 2018, 90, 260–271.
- Shulga, A.; Magalhães, A.C.; Autio, H.; Plantman, S.; di Lieto, A.; Nykjær, A.; Carlstedt, T.; Risling, M.; Arumäe, U.; Castrén, E. The loop diuretic bumetanide blocks posttraumatic p75NTR upregulation and rescues injured neurons. J. Neurosci. 2012, 32, 1757–1770.
- Kravitz, A.V.; Kreitzer, A.C. Striatal mechanisms underlying movement, reinforcement, and punishment. Physiology (Bethesda) 2012, 27, 167–177.
- Simpson, E.H.; Kellendonk, C.; Kandel, E. A possible role for the striatum in the pathogenesis of the cognitive symptoms of schizophrenia. Neuron 2010, 65, 585–596.
- Lemiere, J.; Decruyenaere, M.; Evers-Kiebooms, G.; Vandenbussche, E.; Dom, R. Cognitive changes in patients with Huntington’s disease (HD) and asymptomatic carriers of the HD mutation—A longitudinal follow-up study. J. Neurol. 2004, 251, 935–942.
- Begeti, F.; Schwab, L.C.; Mason, S.L.; Barker, R.A. Hippocampal dysfunction defines disease onset in Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2016, 87, 975–981.
- Paulsen, J.S. Cognitive impairment in Huntington disease: Diagnosis and treatment. Curr. Neurol. Neurosci. Rep. 2011, 11, 474–483.
- Petersen, A.; Gabery, S. Hypothalamic and Limbic System Changes in Huntington’s Disease. J. Huntingt. Dis. 2012, 1, 5–16.
- Arnulf, I.; Nielsen, J.; Lohmann, E.; Schieffer, J.; Wild, E.; Jennum, P.; Konofal, E.; Walker, M.; Oudiette, D.; Tabrizi, S.; et al. Rapid Eye Movement Sleep Disturbances in Huntington Disease. Arch. Neurol. 2008, 65, 482–488.
- Martinez-Horta, S.; Perez-Perez, J.; van Duijn, E.; Fernandez-Bobadilla, R.; Carceller, M.; Pagonabarraga, J.; Pascual-Sedano, B.; Campolongo, A.; Ruiz-Idiago, J.; Sampedro, F.; et al. Neuropsychiatric symptoms are very common in premanifest and early stage Huntington’s Disease. Parkinsonism Relat. Disord. 2016, 25, 58–64.
- Orth, M.; Schippling, S.; Schneider, S.A.; Bhatia, K.P.; Talelli, P.; Tabrizi, S.J.; Rothwell, J.C. Abnormal motor cortex plasticity in premanifest and very early manifest Huntington disease. J. Neurol. Neurosurg. Psychiatry 2010, 81, 267–270.
- Paulsen, J.S.; Langbehn, D.R.; Stout, J.C.; Aylward, E.; Ross, C.A.; Nance, M.; Guttman, M.; Johnson, S.; MacDonald, M.; Beglinger, L.J.; et al. Detection of Huntington’s disease decades before diagnosis: The Predict-HD study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 874–880.
- Bellosta Diago, E.; Perez Perez, J.; Santos Lasaosa, S.; Viloria Alebesque, A.; Martinez Horta, S.; Kulisevsky, J.; Lopez Del Val, J. Circadian rhythm and autonomic dysfunction in presymptomatic and early Huntington’s disease. Parkinsonism Relat. Disord. 2017, 44, 95–100.
- Vonsattel, J.-P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P., Jr. Neuropathological Classification of Huntington’s Disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577.
- Li, J.Y.; Plomann, M.; Brundin, P. Huntington’s disease: A synaptopathy? Trends Mol. Med. 2003, 9, 414–420.
- Tyebji, S.; Hannan, A.J. Synaptopathic mechanisms of neurodegeneration and dementia: Insights from Huntington’s disease. Prog. Neurobiol. 2017, 153, 18–45.
- Garret, M.; Du, Z.; Chazalon, M.; Cho, Y.H.; Baufreton, J. Alteration of GABAergic neurotransmission in Huntington’s disease. CNS Neurosci. Ther. 2018, 24, 292–300.
- Sardini, A.; Amey, J.S.; Weylandt, K.H.; Nobles, M.; Valverde, M.A.; Higgins, C.F. Cell volume regulation and swelling-activated chloride channels. Biochim. Biophys. Acta 2003, 1618, 153–162.
- Wilson, C.S.; Mongin, A.A. The signaling role for chloride in the bidirectional communication between neurons and astrocytes. Neurosci. Lett. 2019, 689, 33–44.
- Duy, P.Q.; David, W.B.; Kahle, K.T. Identification of KCC2 Mutations in Human Epilepsy Suggests Strategies for Therapeutic Transporter Modulation. Front. Cell Neurosci. 2019, 13, 515.
- Elorza-Vidal, X.; Gaitan-Penas, H.; Estevez, R. Chloride Channels in Astrocytes: Structure, Roles in Brain Homeostasis and Implications in Disease. Int. J. Mol. Sci. 2019, 20, 1034.
- Deeb, T.Z.; Lee, H.H.; Walker, J.A.; Davies, P.A.; Moss, S.J. Hyperpolarizing GABAergic transmission depends on KCC2 function and membrane potential. Channels (Austin) 2011, 5, 475–481.
- Hübner, C.A.; Stein, V.; Hermans-Borgmeyer, I.; Meyer, T.; Ballanyi, K.; Jentsch, T.J. Disruption of KCC2 Reveals an Essential Role of K-Cl Cotransport Already in Early Synaptic Inhibition. Neuron 2001, 30, 515–524.
- Huebner, C.A.; Holthoff, K. Anion transport and GABA signaling. Front. Cell. Neurosci. 2013, 7, 177.
- Staley, K.J.; Soldo, B.L.; Proctor, W.R. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science 1995, 269, 977–981.
- Dallwig, R.; Deitmer, J.W.; Backus, K.H. On the mechanism of GABA-induced currents in cultured rat cortical neurons. Pflügers Arch. 1999, 437, 289–297.
- Kim, D.-Y.; Fenoglio, K.A.; Kerrigan, J.F.; Rho, J.M. Bicarbonate contributes to GABAA receptor-mediated neuronal excitation in surgically resected human hypothalamic hamartomas. Epilepsy Res. 2009, 83, 89–93.
- Ma, B.F.; Xie, M.J.; Zhou, M. Bicarbonate efflux via GABAA receptors depolarizes membrane potential and inhibits two-pore domain potassium channels of astrocytes in rat hippocampal slices. Glia 2012, 60, 1761–1772.
- Lombardi, A.; Jedlicka, P.; Luhmann, H.J.; Kilb, W. Interactions between membrane resistance, GABA-A receptor properties, bicarbonate dynamics and Cl−-transport shape activity-dependent changes of intracellular Cl− concentration. Int. J. Mol. Sci. 2019, 20, 1416.
- Sibbe, M.; Kulik, A. GABAergic regulation of adult hippocampal neurogenesis. Mol. Neurobiol. 2017, 54, 5497–5510.
- Kim, Y.; Jun, I.; Shin, D.H.; Yoon, J.G.; Piao, H.; Jung, J.; Park, H.W.; Cheng, M.H.; Bahar, I.; Whitcomb, D.C.; et al. Regulation of CFTR Bicarbonate Channel Activity by WNK1: Implications for Pancreatitis and CFTR-related disorders. Cell Mol. Gastroenterol. Hepatol. 2019.
- Chiu, C.Q.; Barberis, A.; Higley, M.J. Preserving the balance: Diverse forms of long-term GABAergic synaptic plasticity. Nat. Rev. Neurosci. 2019, 20, 272–281.
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255.
- Medina, I.; Friedel, P.; Rivera, C.; Kahle, K.T.; Kourdougli, N.; Uvarov, P.; Pellegrino, C. Current view on the functional regulation of the neuronal K+-Cl− cotransporter KCC2. Front. Cell Neurosci. 2014, 8, 27.
- Singh Jaggi, A.; Kaur, A.; Bali, A.; Singh, N. Expanding spectrum of sodium potassium chloride co-transporters in the pathophysiology of diseases. Curr. Neuropharmacol. 2015, 13, 369–388.
- Smith, K.R.; Muir, J.; Rao, Y.; Browarski, M.; Gruenig, M.C.; Sheehan, D.F.; Haucke, V.; Kittler, J.T. Stabilization of GABAA receptors at endocytic zones is mediated by an AP2 binding motif within the GABAA receptor β3 subunit. J. Neurosci. 2012, 32, 2485–2498.
- Plotkin, M.D.; Snyder, E.Y.; Hebert, S.C.; Delpire, E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: A possible mechanism underlying GABA′s excitatory role in immature brain. J. Neurobiol. 1997, 33, 781–795.
- Yamada, J.; Okabe, A.; Toyoda, H.; Kilb, W.; Luhmann, H.J.; Fukuda, A. Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J. Physiol. 2004, 557, 829–841.
- Deng, X.; Dzamko, N.; Prescott, A.; Davies, P.; Liu, Q.; Yang, Q.; Lee, J.D.; Patricelli, M.P.; Nomanbhoy, T.K.; Alessi, D.R.; et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol. 2011, 7, 203–205.
- Huang, H.; Song, S.; Banerjee, S.; Jiang, T.; Zhang, J.; Kahle, K.T.; Sun, D.; Zhang, Z. The WNK-SPAK/OSR1 Kinases and the Cation-Chloride Cotransporters as Therapeutic Targets for Neurological Diseases. Aging Dis. 2019, 10, 626–636.
- Alessi, D.R.; Zhang, J.; Khanna, A.; Hochdorfer, T.; Shang, Y.; Kahle, K.T. The WNK-SPAK/OSR1 pathway: Master regulator of cation-chloride cotransporters. Sci. Signal. 2014, 7, re3.
- Gamba, G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 2005, 85, 423–493.
- Orlov, S.N.; Koltsova, S.V.; Kapilevich, L.V.; Gusakova, S.V.; Dulin, N.O. NKCC1 and NKCC2: The pathogenetic role of cation-chloride cotransporters in hypertension. Genes Dis. 2015, 2, 186–196.
- Nezu, A.; Parvin, M.N.; Turner, R.J. A conserved hydrophobic tetrad near the C terminus of the secretory Na+-K+-2Cl− cotransporter (NKCC1) is required for its correct intracellular processing. J. Biol. Chem. 2009, 284, 6869–6876.
- Chew, T.A.; Orlando, B.J.; Zhang, J.; Latorraca, N.R.; Wang, A.; Hollingsworth, S.A.; Chen, D.-H.; Dror, R.O.; Liao, M.; Feng, L. Structure and mechanism of the cation–chloride cotransporter NKCC1. Nature 2019, 572, 488–492.
- Hsu, Y.T.; Chang, Y.G.; Liu, Y.C.; Wang, K.Y.; Chen, H.M.; Lee, D.J.; Yang, S.S.; Tsai, C.H.; Lien, C.C.; Chern, Y. Enhanced Na+-K+-2Cl− 1 underlies motor dysfunction in huntington’s disease. Mov. Disord. 2019, 34, 845–857.
- Dargaei, Z.; Bang, J.Y.; Mahadevan, V.; Khademullah, C.S.; Bedard, S.; Parfitt, G.M.; Kim, J.C.; Woodin, M.A. Restoring GABAergic inhibition rescues memory deficits in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA 2018, 115, E1618–E1626.
- Tang, B.L. The Expanding Therapeutic Potential of Neuronal KCC2. Cells 2020, 9, 240.
- Moore, Y.E.; Kelley, M.R.; Brandon, N.J.; Deeb, T.Z.; Moss, S.J. Seizing Control of KCC2: A New Therapeutic Target for Epilepsy. Trends Neurosci. 2017, 40, 555–571.
- Kahle, K.T.; Khanna, A.R.; Alper, S.L.; Adragna, N.C.; Lauf, P.K.; Sun, D.; Delpire, E. K-Cl cotransporters, cell volume homeostasis, and neurological disease. Trends Mol. Med. 2015, 21, 513–523.
- Tillman, L.; Zhang, J. Crossing the Chloride Channel: The Current and Potential Therapeutic Value of the Neuronal K+–Cl− Cotransporter KCC2. Biomed. Res. Int. 2019, 2019, 8941046.
- de Los Heros, P.; Alessi, D.R.; Gourlay, R.; Campbell, D.G.; Deak, M.; Macartney, T.J.; Kahle, K.T.; Zhang, J. The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+–Cl− co-transporters. Biochem. J. 2014, 458, 559–573.
- Lu, D.C.-Y.; Hannemann, A.; Wadud, R.; Rees, D.C.; Brewin, J.N.; Low, P.S.; Gibson, J.S. The role of WNK in modulation of KCl cotransport activity in red cells from normal individuals and patients with sickle cell anaemia. Pflug. Arch. Eur. J. Phy. 2019, 471, 1539–1549.
- Shekarabi, M.; Zhang, J.; Khanna, A.R.; Ellison, D.H.; Delpire, E.; Kahle, K.T. WNK Kinase Signaling in Ion Homeostasis and Human Disease. Cell Metab. 2017, 25, 285–299.
- Heubl, M.; Zhang, J.; Pressey, J.C.; Al Awabdh, S.; Renner, M.; Gomez-Castro, F.; Moutkine, I.; Eugene, E.; Russeau, M.; Kahle, K.T.; et al. GABAA receptor dependent synaptic inhibition rapidly tunes KCC2 activity via the Cl−-sensitive WNK1 kinase. Nat. Commun. 2017, 8, 1776.
- Brown, A.; Meor Azlan, N.F.; Wu, Z.; Zhang, J. WNK-SPAK/OSR1-NCC kinase signaling pathway as a novel target for the treatment of salt-sensitive hypertension. Acta Pharm. Sin. 2020.
- Meor Azlan, N.F.; Zhang, J. Role of the Cation-chloride-cotransporters in Cardiovascular Disease. Cells 2020, 9, 2293.
- Kahle, K.T.; Khanna, A.R.; Duan, J.; Staley, K.J.; Delpire, E.; Poduri, A. The KCC2 Cotransporter and Human Epilepsy: Getting Excited About Inhibition. Neuroscientist 2016, 22, 555–562.
- Yeo, M.; Berglund, K.; Augustine, G.; Liedtke, W. Novel repression of Kcc2 transcription by REST-RE-1 controls developmental switch in neuronal chloride. J. Neurosci. 2009, 29, 14652–14662.
- Smith-Dijak, A.I.; Sepers, M.D.; Raymond, L.A. Alterations in synaptic function and plasticity in Huntington disease. J. Neurochem. 2019, 150, 346–365.
- Rinehart, J.; Maksimova, Y.D.; Tanis, J.E.; Stone, K.L.; Hodson, C.A.; Zhang, J.; Risinger, M.; Pan, W.; Wu, D.; Colangelo, C.M.; et al. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 2009, 138, 525–536.
- Friedel, P.; Kahle, K.T.; Zhang, J.; Hertz, N.; Pisella, L.I.; Buhler, E.; Schaller, F.; Duan, J.; Khanna, A.R.; Bishop, P.N.; et al. WNK1-regulated inhibitory phosphorylation of the KCC2 cotransporter maintains the depolarizing action of GABA in immature neurons. Sci. Signal. 2015, 8, ra65.
- Watanabe, M.; Zhang, J.; Mansuri, M.S.; Duan, J.; Karimy, J.K.; Delpire, E.; Alper, S.L.; Lifton, R.P.; Fukuda, A.; Kahle, K.T. Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival. Sci. Signal. 2019, 12, eaaw9315.
- Moore, Y.E.; Conway, L.C.; Wobst, H.J.; Brandon, N.J.; Deeb, T.Z.; Moss, S.J. Developmental Regulation of KCC2 Phosphorylation Has Long-Term Impacts on Cognitive Function. Front. Mol. Neurosci. 2019, 12, 173.