Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Esmaa Bouhamida and Version 4 by Jessie Wu.
Heart valve diseases are a major contributor to cardiovascular morbidity and mortality worldwide. They affect more than 13% of the population aged over 75 years old and occur when any type of the four heart valves (tricuspid, pulmonic, mitral, and aortic valves) is damaged. Calcific aortic valve disease (CAVD) is defined as a slowly progressing condition that ranges from mild valve aortic sclerosis to severe calcifying aortic valve stenosis. This progression manifests in approximately 2% of individuals over 65 years old annually.
- calcific aortic valve stenosis
- hypoxia
- HIF-1α
- mitochondria
- oxidative stress
- inflammation
- therapeutic target
1. Hypoxia Signaling and Molecular Regulation of Hypoxia-Inducible Factor F-1
Hypoxia plays a critical role in cardiovascular diseases (CVDs) and is orchestrated by a hallmark heterodimer trans-acting DNA-binding hypoxia-inducible transcription factors (HIFs), which are key regulators mediating adaptation to hypoxic conditions and are modulated by an O2-sensitive-expressed alpha subunit (HIF-1α) (or its analogs HIF-2α and HIF-3α). In normal conditions, HIF-1α is continuously synthesized and hydroxylated through HIF prolyl-4-hydroxylases, leading to its rapid ubiquitination and proteasomal degradation (ubiquitin proteasome 26S), the von Hippel–Lindau (pVHL) function as a tumor suppressor binds to the ubiquitin ligase complex E3 targeting the HIF-1α subunit destruction in the O2 degradation domain, causing its short life. In contrast, under hypoxia, HIF-1α hydroxylation is suppressed through the inhibition of the O2-dependent propyl-hydroxylase-1, -2, and -3 enzyme activity (PHD1, -2, and -3), leading to the stabilization of HIF-1α in the cytosol, and migrates to the nucleus, where it forms a heterodimer with the beta subunits (HIF-1β, aryl hydrocarbon receptor nuclear translocator, ARNT) that bind to a core putative regulatory sequence called hypoxia response elements (HRE) with a consensus sequence (5′-RCGTG-3′) in the promoter or enhancer of target genes to enhance a concerted transcriptional response during a hypoxic condition [1][35] (Figure 1). Both α and β subunits have basic helix-loop-helix (bHLH) motifs, a DNA-binding domain that can bind HREs to target specific genes [2][3][8,36]. The transcription activity of the target genes requires not only the transfer of HIF-1α to the nucleus but also the complex HIF-1 requires the recruitment of multiple cofactors such as CREB-binding protein (CBP)/p300 and transcription intermediary factor 2 steroid-receptor activator that binds to the CTAD domain, and another cofactor that increases the HIF-1/HRE complex binding the M2 isoform of pyruvate kinase (PKM2) [4][37]. The canonical sensor of hypoxia, HIF-1α, mediates a cellular response during hypoxic conditions through the regulation of the transcription activity of enormous target genes, termed hypoxia-inducible genes encoding proteins, as examples: the lactate dehydrogenase-A (LDH-A) or pyruvate dehydrogenase kinase isoform 1 (PDK) [5][6][38,39]; VEGF-A [7][40]; erythropoietin (EPO) [8][41]; and inducible nitric oxide synthase (iNOS) [9][42], which are needed for improving tissue O2 homeostasis, energy metabolism, and efficient management of hypoxia-induced toxic stress, and elicit a crucial impact in various CVDs, such as ischemic heart disease (IHD) and HF [10][11][7,43]. HIF-2α, the analog of HIF-1α, is also termed endothelial PAS domain protein-1 (EPAS-1), and is predominantly enriched within endothelial cells and in highly vascularized tissues [12][44]. These two subunits (HIF-1α and HIF-2α) contain different spatial expressions in IHD, for example, and various effects in response to hypoxia [13][45]. Despite the recent interest in studying hypoxia and HIF-1 in CVDs, their role at the onset of CAVS remains unclear and requires deep further investigation.
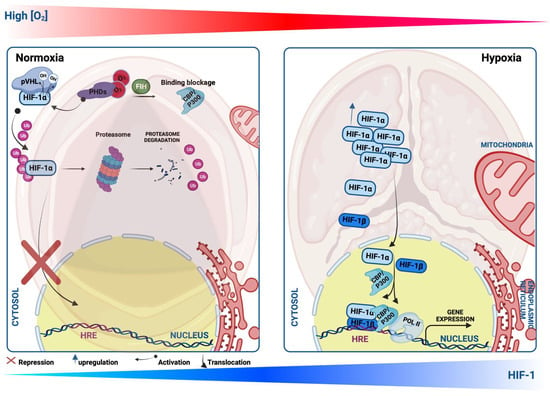
Figure 1. Schematic illustration showing Hypoxia-inducible factor-1 α (HIF-1α) protein regulation during normoxia and hypoxia. Under the normoxic condition, HIF-1α protein is hydroxylated by propyl-hydroxylases (PHDs) and factor-inhibiting HIF (FIH), which facilitate the binding of HIF-1α with the von Hippel–Lindau protein (pVHL), leading to its ubiquitination, and thus proteasomal degradation. Upon the PHD inhibition or hypoxic condition, HIF-1α translocates to the nucleus, where it heterodimerizes with HIF-1β and binds to a core putative sequences of target genes termed hypoxia response element (HRE) and stimulates their transcriptional activity.
2. Hypoxia Signaling and Inflammation in Calcific Aortic Valve VDisease
The heart AV is an avascular tissue able to sustain metabolic activity, nutrition, and oxygenation through passive diffusion. Nevertheless, with age, the initiation of CAVS is exhibited by endothelial injury triggered by shear stress, lipid deposition, and inflammation [14][46]. In these early stages of CAVS, the thickening of the valve process compromises the diffusion of O2, resulting in tissue hypoxia, while the CAVS progression occurs through the abnormal remodeling of the ECM, which is modulated by the valve interstitial cells [15][16][47,48]. Sustained stimulation of hypoxia maintains HIF-1α signaling, leading to the upregulation of inflammation and fibrosis, but these effects are counterbalanced by sustained HIF-2α signaling, potentially linked to mitochondrial and peroxisomal abnormalities [17][49]. It is demonstrated that during hypoxia, HIF-1α and HIF-2α are activated and both can transactivate VEGF expression [18][19][20][50,51,52]. HIF-1α is reported to initiate the process of angiogenesis, while HIF-2α is required for vascular network maturation [21][22][23][53,54,55]. Nevertheless, it is not yet established whether HIF-2α contributes to the angiogenesis maturation and regenerative process in CAVS.
Hypoxia has been recently identified in both the aortic and mitral valves [24][25][56,57]. Moreover, diseased valvular interstitial cells in regions surrounding calcific nodules have been found to express HIF-1α, which is a pro-angiogenic transcription factor [26][27][9,10]. Additionally, in human aortic endothelial cells (HAVEC), the level of HIF-1α mRNA and protein expression are identified as elevated in response to disturbed flow as opposed to stable flow [28][58]. A study has demonstrated the significant upregulation of HIF-1α in the stenotic valves and its colocalization with angiogenesis in areas calcified [27][10]. These findings suggest a further key role of HIF-1α in influencing the valve phenotype through the induction of VEGF expression, thereby activating neo-angiogenesis, a feature of valvular disorders, to increase metabolic adaptation [27][29][10,59]. In contrast to the upregulation of pro-angiogenic factors, anti-angiogenic factors have been identified as suppressed [30][31][32][60,61,62]. Furthermore, there are five VEGF family growth factors that bind to specific tyrosine kinase receptors and play important roles in the formation of new blood vessels and lymphatic vessels [33][34][63,64]. Among the VEGF receptors is the soluble fms-like tyrosine kinase 1 (sFlt1), an anti-angiogenic component that sustains AV avascularity [35][65].
A recent study by Lewis and colleagues has shown that sFlt1 is expressed in the native normal AV, but its expression level is significantly downregulated in patients with CAVS [35][65]. The authors demonstrated for the first time the dual roles of hypoxia in stimulating angiogenesis in CAVS, as the classical way by inducing VEGF-A and by inhibiting the sFlt1 expression, which could elevate inflammation, thus contributing to CAVS progression [35][65]. Sphingosine 1-phosphate (SP1) is a bioactive lipid signaling mediator shown to inhibit angiogenesis via the activation of sFlt1 expression in CAVS patients [35][65]. However, the link between HIFs (HIF-1α and HIF-2α) and sFlt1 has not been studied yet in CAVS, and its mechanism of action in response to hypoxia is not clear. To our knowledge, the relationship between sFlt1 and HIFs in response to hypoxia has been studied only in the pathogenesis of Preeclampsia, the onset of hypertension during pregnancy [36][37][66,67]. The relationship between HIF-1 and sFlt1 needs to be studied to understand further the molecular mechanisms behind this process in CAVS. Prior studies have shown that sFlt1 induces inflammation in VICs when it synergizes with lipopolysaccharide (LPS), which is a major component of the outer membrane of Gram-negative bacteria [38][68]. LPS drives the activation of Toll-like receptor 4 (TLR4), a key receptor of innate and adaptive immunity, stimulating the inflammatory (interleukin 6 and 8 (IL-6, IL-8), and intercellular adhesion molecule-1 (ICAM-1)) and osteogenic responses (bone morphogenetic protein-2 (BMP-2) and runt-related transcription factor 2 (RUNX2) in CAVS patients [39][40][69,70]. IL-37 is a novel cytokine member of the IL-1 family and plays a potential role in suppressing inflammatory responses, identified as downregulated in CAVS [41][42][71,72]. The lower levels of IL-37 observed in CAVS patients have been explained by M1 macrophage infiltration in pathological AV stenosis (AVs). IL-37 inhibits the macrophages polarization M1 (reduction in IL-6, MCP-1, iNOS, and the surface marker of M1 (CD11c)) via the suppression of nuclear factor kappa B (NF-κB) and Notch homolog 1, translocation-associated (Notch1) signaling pathways [43][73]. Along with that, IL-37 is reported as a potent anti-osteogenic in AVICs from patients, through the suppression of the NF-κB and extracellular signal-regulated kinase (ERK1/2) [42][72]. The anti-inflammatory response of IL-37 is associated with NF-κB, suppressing the TLR4 ligand LPS-mediated IL-6, IL-8, monocyte chemoattractant protein-1 (MCP-1), and ICAM-1 stimulation in AVICs from patients [44][74], thus playing a critical role in CAVS physiopathology. Noteworthy, IL-37 is capable of inducing other anti-inflammatory pathways including the AMP-activated protein kinase (AMPK) and Phosphatase and tensin homolog, which may impact NF-κB stimulation in CAVS [41][71]. Morciano and colleagues have shown the correlation of both pro-inflammatory cytokine levels of IL-18 and IL-1β—which both belong to the IL-1 family—in CAVS [14][46]. Furthermore, IL-1β has been found to stimulate mTORC1, and, downstream, it enhances HIF-1α activity in several diseases [45][75]. The expression of the HIF-1α protein is induced by IL-1β in normoxia in multiple cell types [46][47][48][76,77,78]. For instance, HIF-2α is one among the different signaling pathways that are involved in M1 infiltration [49][79], but is yet to be studied in macrophages of AVICs. Previous studies have shed light on better understanding the further relationship between HIF-1, Signal Transducer and Activator of Transcription 3 (STAT3), and IL-37 in cancer disorders [50][80]. This would increase further interest in a deep understanding of the crosstalk of the IL-37 and HIF-1 in the pathogenesis of CAVS. Evidence has confirmed the upregulation of HIF-1α and its analog HIF-2α in valve stenosis. Interestingly, HIF-2α co-localizes with NF-κB in regions of calcified lesions of AS of patients [20][52]. These findings have been correlated positively with the enhanced levels of VEGF and the formation of neovessels [20][52]. On the other hand, it is well known that the expression of VEGF can also be increased by inflammatory cytokines [51][81]. The findings indicate further convergence between hypoxia and inflammatory mechanisms involved in the remodeling of the valve ECM, which contributes to VIC stimulation and calcification. It has been shown that the HIF-1α activates the expression of various proteins involved in ECM remodeling including the Neutrophil Gelatinase-Associated Lipocalin [52][53][82,83] and the MMP2 and MMP9, suggesting the key impact of hypoxia in ECM remodeling.
In light of the previous findings, a novel immune non-hypoxic process entailing the combination between LPS and interferon-γ (IFN-γ) has been explored to stimulate calcification in AVICs from patients, through the STAT1/HIF-1α signaling pathway [54][84]. Interestingly, this response to HIF-1α stimulation mediated by LPS is sex-dependent, as it is more robust in VICs from male donors compared to females [54][84]. The Janus kinases (JAK)-STAT signaling pathways are hallmark regulatory routes contributing to several cytokine responses activating mineralization and calcification of AV interstitial tissues, including IL-6 and IFN-γ [54][55][56][84,85,86]. Notably, the study of Parra-Izquierdo and colleagues also suggests further a relationship between JAK-STAT and HIF-1α-dependent sex differences in the context of CAVS [54][84]. However, females display lower responses and tend to be more protective compared with males due to the activation of the phosphatidylinositol 3-kinase (PI3K)-AKT signaling survival pathways [57][87]. Nevertheless, more research is needed to fully understand these relationships.
These findings propose the further potential involvement of HIF-1α in normoxia and in the early phases of CAVS when the hypoxic event is not yet activated; however, it is not known how LPS and/or IFN-γ could stabilize HIF-1α. Since the stabilization of HIF-1α is regulated by prolyl-4-hydroxylases under normoxic conditions, it is therefore possible that LPS and/or IFN-γ treatment could cause a decrease in hydroxylation, leading to the activation of HIF-1α. Other pioneer evidence supports the upregulation of HIF-1α in normoxic conditions—mediating the calcification process in AVICs. The ubiquitin E2 ligase C (UBE2C) is a member of the Anaphase Promoting Complex/Cyclosome (APC/C), which has been reported to also bind pVHL [58][88]. UBE2C upregulates the endothelial–mesenchymal transition (EndMT) and endothelial AV inflammation via the stimulation of HIF-1α levels through further ubiquitination and degradation of its upstream modulator pVHL, and this was accompanied by the reduction in microRNA-483–3p (miR-483) in HAECs [28][58]. In addition, the miR-483 mimics and the pharmacological suppressor of HIF-1α (PX478) significantly downregulate the porcine AV calcification through UBE2C reduction [28][58]. Other evidence supports the involvement of HIF-1α in valve calcification: PX478 significantly blocked the deposition of calcium resulting from distributed flow, and the response was more effective in male valve interstitial cells [27][59][10,11]. Other in vitro studies have identified the increased expression levels of HIF-1 signaling pathways including IL-6, HIF-1α, and Heme Oxygenase 1 (HMOX1) in CAVS-related ferroptosis signaling pathways [60][89]. Hence, one of the causes of ferroptosis is an iron overload that is involved in CAVS by enhancing calcium deposition and calcification in endothelial cells (HUVEC) [61][90]. It is identified that the endothelial cells play crucial impacts in the calcification process through the EndMT [62][91]. Multiple pathways are involved in this process including the signaling pathways involved in hypoxia and inflammation, such as the transforming growth factor beta (TGF-β) signaling pathway [63][92], and the Wnt signaling pathway [64][65][93,94].
In the same regard, inflammation is also triggered by fetuin-A (alpha2-Heremans Schmid glycoprotein); a 59 kDa glycoprotein synthesized in the liver, emerges as a potent circulating inhibitor of the calcification process, modulates macrophage polarization, and attenuates inflammation and fibrosis [66][95]. Intracellular fetuin-A suppresses calcification stimulated by transforming growth factor-β and bone morphogenetic proteins [67][68][96,97]. In addition, previous works have conflicting results on circulating fetuin-A as a biomarker for CAVD. Nevertheless, a meta-analysis demonstrated significantly lower levels of fetuin-A in AS patients compared to healthy conditions; also, fetuin-A levels have been significantly identified as being associated with CVD risk factors including age, male gender, smoking, low-density lipoprotein (LDL) and TG, hypertension, and diabetes [69][98]. Studies have shown an inverse correlation between fetuin-A levels and the progression of calcific AV and underlined diminished fetuin-A levels in AV sclerosis patients, the early asymptomatic phase of CAVD, suggesting that fetuin-A is an early calcification biomarker [70][71][99,100]. These studies suggest further involvement of fetuin-A in the initiation of AV calcification, hence raising the concept that fetuin-A, as an inhibitor of calcification, may prevent valvular calcifications when the calcium phosphate is disrupted, suggesting its correlation to calcium tissue deposition [72][101], and its levels, is also associated with inflammation and other comorbidities such as chronic kidney disease and diabetes [73][102]. In a very recent study, Chen et al. demonstrated the involvement of Fetuin-A in calcific osteogenic environment-induced VICs calcification, and in parallel, its level reported the decreased inhibition of miR-101 taking place [74][103]. However, the role of fetuin-A in the progression of CAVD has not been clearly investigated. Therefore, relationships of fetuin-A with tissue calcification and cardiovascular diseases in general are divergent, reflecting its diverse action. Notably, beyond the role of fetuin-A as a calcification suppressor in the serum phase, it acts as a potent calcium mineral scavenger, preventing the ectopic pathological calcification of the tissue especially in response to hypoxic stress in renal tissue remodeling upon IH injury [66][95]. Recently, fetuin-A has been identified as an evolutionary target gene of HIF-1 [75][104]; however, the correlation between HIF-1 and fetuin-A is still unclear in CAVS. Further understanding the complex interplay between HIF-1, fetuin-A, and pro-inflammatory cytokine in CAVS patients would pave the way to better predict the presence of CAVS and may provide further molecular targeting strategies.
3. Mitochondrial Impairment in Calcific Aortic Valve VDisease
Mitochondria have key roles in eukaryotic cells, as they control several cellular mechanisms such as bioenergetics, signal transduction, and energy metabolism. Furthermore, these organelles are major regulators and executioners of cell death mechanisms, in particular autophagy and apoptosis [76][105]. To regulate these processes, cells have to preserve a functional mitochondrial population, a fundamental aspect allowed by the mitochondrial quality control system, in which mitochondrial dynamics, mitochondrial biogenesis, and mitochondrial autophagy (named mitophagy) are the main events [77][106]. Fusion and fission help to keep mitochondrial structure integrity and are necessary steps to improve metabolism and facilitate cooperation and communication between mitochondria [78][107]. At the same time, erroneous fission and fusion events may provoke the formation of a nonfunctional mitochondria population, which can induce the production of damaged elements (such as mitochondrial reactive oxygen species) that are harmful for the entire mitochondria population and for the cell. Injured mitochondria can be removed by the degradative process, mitophagy, which recognizes and sequesters the damaged organelle into autophagy vesicles, which are then delivered to the lysosome for degradation [79][108]. Finally, preserving the adequate mitochondrial number of the cell may intervene in mitochondrial biogenesis that is responsible for generating new mitochondrial offspring [80][109]. Decline and/or sustained activation of these molecular mechanisms deputed to control the mitochondrial amount and quality has been associated with several human diseases, in particular, mitochondrial diseases [81][110], genetic disorders [82][111], neurodegeneration [83][84][112,113], cancer [85][114], and cardiovascular disorders [14][46].
For instance, immunostained calcified human AVs revealed high levels of the mitochondrial fission initiator protein dynamin-related protein 1 (DRP1). Inhibition of DRP1 via RNA interference promotes a reduction in the osteogenic differentiation process and inhibits oxidative stress [86][115]. A more recent study highlighted the overexpression of protein tyrosine phosphatase 1B (PTP1B) in CAVD [87][116]. PTP1B is a negative regulator of the leptin and insulin signaling pathways, which are involved in the regulation of mitochondrial dynamics and biogenesis [88][89][117,118]. The authors demonstrate the decreased osteogenic differentiation of interstitial valvular cells by the pharmacological inhibition of PTPB1. This effect is accompanied by an upregulation of mitochondrial biogenesis, which has been observed to be downregulated during the progression of valvular calcification [87][116]. Morciano et al. revealed the presence of aged mitochondria together with reduced PGC-1α expression, a key mitochondrial biogenesis protein, in interstitial cells isolated from human patients [14][46]. Impairment of mitochondrial biogenesis in CAVS is associated with increased cell death and the presence of aged mitochondria, despite an increase in mitophagy and autophagy fluxes, suggesting an insufficient turnover of mitochondria in CAVD samples. Moreover, the authors revealed for the first time the presence of other mitochondrial impairments in CAVD patients, such as calcium dysregulation, reduced respiratory capacity, and lack of ATP production [14][46].
The gene expression profile of AV tissue identified several novel genes associated with mitochondrial functions variations that are involved in the pathogenesis of CAVD, such as an increase in reactive oxygen species (ROS) production and reduced mitochondrial membrane potential, metabolic imbalance, and mitochondrial fragmentation [90][119]. Moreover, the authors suggest that integrated miRNA/mRNA analyses might be used as diagnostic biomarkers for CAVD [90][119]. Interestingly, they later identified matrix metalloproteinase 9 expression (MMP9), a mitochondrial-related gene, as being extremely high in AS samples, which would be a useful biomarker for aortic stenosis [91][120].
Inside the cell, Calcium (Ca2+) is one of the major second messengers and regulates a plethora of biological processes, such as metabolism, antioxidant defense, apoptosis, muscle contraction, neurotransmitter release, and also mitochondrial functioning [92][121]. Therefore, it is not surprising that dysregulations of Ca2+ dynamics are involved in different pathologies [93][122]. In the context of aortic calcification, evidence that connects aortic stenosis and Ca2+ dysregulation goes back to a genome-wide association study [94][123], where calcified valves reveal the upregulation of mRNA levels of RUNX2 concomitant with an increased calcium voltage-gated channel subunit alpha 1 C (CACNA1C) gene [94][123], which encodes the CaV1.2 L-type voltage-gated Ca2+ channel. Later, these data were confirmed by another study, which demonstrated a higher Ca2+ influx in CAVD patients through the CaV1.2 channel [95][124].
VEGF is a target gene of HIF-1α that regulates several cellular processes including proliferation, cell survival, differentiation, and migration [96][125]. Xu et al. have demonstrated the impact of VEGF in sustaining the mitochondrial fission and fusion balance, mitigating the mitochondrial apoptotic pathway; thereby, VEGF could play a critical role in repairing the AS transition from compensatory cardiac hypertrophy to HF in mouse animal models [96][125].
It is important to note that patients with AS exhibit a metabolic shift from fatty acid to glucose metabolism, which is characterized by a decreased expression of fatty acid translocase (FAT/CD36) protein, together with a downregulation of other fatty acid transporters, such as plasma membrane and heart-type cytosolic fatty acid binding proteins (FABPpm and H-FABP), β-oxidation, Krebs cycle, and oxidative phosphorylation proteins. On the contrary, the same cardiac biopsy exhibits an increased expression of glucose transporter 1 and 4 (Glut 1, 4). This research tudy suggests a downregulation of fatty acid oxidation proportionally to a more severe outcome in patients with aortic stenosis [97][126]. Nonetheless, whether this metabolic shift in CAVS patients results from the involvement of mitochondrial ROS (mtROS) in instigating AV calcification or whether another mediator contributes to this process remains unknown.
Notably, such a metabolic shift to glycolysis is well demonstrated in various CVDs orchestrated by HIF-1α; however, its effect on the metabolism adaptation in CAVD is yet to be explored. HIF-1α regulates multiple genes influencing mitochondrial activity and is crucial for the metabolic shift, such as LDH-A and phosphoglycerate kinase-1 (PGK1) [98][127], thus elevating anaerobic glycolysis by enhancing the generation of glycolysis enzymes, increasing glucose transporters expression (e.g., Glut 1, 4), and inhibiting mitochondrial energy metabolism [99][100][128,129], while impeding fatty acid oxidation [10][101][7,130]. Several studies have supported the notion that HIF-1α plays a significant role in oxidative stress, as its expression level is intrinsically associated with mtROS in response to O2 deprivation [10][102][7,131].
Furthermore, it has been reported that human calcified valves are enriched in oxidative stress, worsening the progression of calcification [103][132]. The increased oxidative stress levels seem to be inversely proportional to antioxidant enzyme expression and functionality, as well as the uncoupling nitric oxide synthase (NOS) activation [104][105][133,134]. Together these data illustrate the crucial role of mitochondria in the pathophysiology of CAVD.
4. Relationship between Inflammation and Mitochondria in Calcific Aortic Valve VDisease
TAs we reviewed in the previous paragraphs, the inflammatory response has been the only known mediator of CAVD in the past [106][107][135,136], and its connection with the onset and progression of the disease that resulted in either surgical valve replacement or transcatheter AV implantation has been extensively reported [108][109][26,137]. Indeed, a significant amount of clinical data, extrapolated from patients affected by AS, showed with different techniques, high levels of inflammation. In the 1990s, aortic valvular lesions from this cohort of patients were carefully analyzed through popular biochemical approaches; the native tissue appeared as being characterized by thickening, by a large amount of lipids deposition, and by the presence of mineralization and calcium accumulation into the leaflet (Figure 2). Concurrently, a high grade of inflammatory infiltrate with foam cell macrophages was present in the lesions [110][138]. From plasma samples of AS patients, IL-1β and IL-18 were the main circulating cytokines to be overexpressed [14][46]; however, IL-6 and TNF have also been reported to play a role in osteogenic differentiation with a significant pool of M1 polarized macrophages [111][139]. In 2012, Dweck MR and co-workers used for the first time the positron emission tomography (PET) and two common PET tracers, 18F-Flurodeoxyglucose (18F-FDG) and 18F-Sodium fluoride (18F-NaF), which were able to target calcification and inflammation into the same patient [112][140]. They found the inflammatory response increased by 91% in AS patients compared to healthy subjects [112][140].
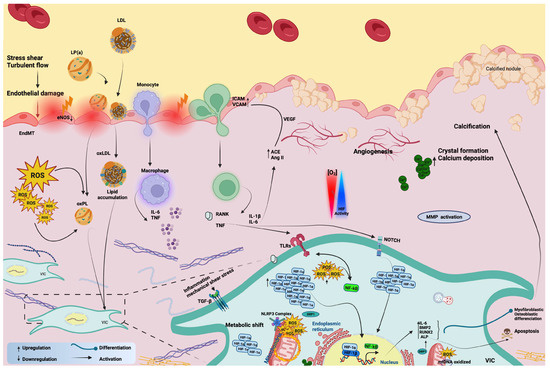
Figure 2.
A diagram representing the contribution of inflammation, mitochondrial dysfunction, and hypoxia signaling in the pathophysiology of the calcific aortic valve.
Today, inflammation is still considered to be the main active player in the phases that precede calcification, but it is deeply nestled among other major contributors such as mitochondrial dysfunctions for which cause–effect relationships are difficult to address [14][108][113][114][26,46,141,142].
The link between mitochondria and inflammation is very close. A seminal paper by Zhong Z. and co-workers showed how mitochondria directly orchestrate signals from TLR to produce oxidized mtDNA fragments, needed to activate NLRP3 inflammasomes once they have moved into the cytosol [115][143]. Basically, according to this evidence, mitochondria constitute a reservoir of many signaling substances. A pool of these, once released into the cytosol, becomes harmful and can trigger mitochondria-mediated inflammation through multiple receptor-dependent responses [116][144]. These are the so-called mitochondrial danger-associated molecular patterns (mtDAMPS) and include, for example, ATP, mtDNA, and mtROS.
NLRP3 is a multiprotein platform that is activated by mtDAMPS; it relocalizes to mitochondria and associated membranes and plays a crucial role in CAVD through the production of the mature form of IL-1β [117][145]. This cytokine is consistently upregulated in CAVD patients, is highly expressed in situ in calcified areas, and further sustains inflammation, stimulating IL-6 and IL-8 production and activating the NF-kB pathway [118][146]. It is documented that IL-1β activates HIF-1α to stimulate a metabolic shift from oxidative phosphorylation (OXPHOS) to glycolysis, which is crucial in adaptive immunity [119][147]. The exacerbation of the inflammatory phenotype is also given by the enhancement of the MPP1 function with drastic changes in the extracellular matrix composition [120][148]. Accordingly, the antagonization of the IL-1 receptor by the IL-1 receptor antagonist (Ra) has been shown to block the proinflammatory pathway [121][149], conferring on the receptor properties targeted for cardioprotection. Moreover, its genetic depletion in animal models of CAVD has definitively shown its crucial contribution to the calcification process of the AV [121][149].
Additionally, to the aforementioned studies in the previous sections, HIF-1α upregulates mtROS levels, stimulates the NF-kB transcription factor, and activates inflammasome genes expression, including NOD-, LRR-, and pyrin domain-containing protein (NLRC)4, NLRP3, and the IL-1β genes. Thereby, leading to mitochondrial oxidative stress, affecting in return mitochondrial membrane permeability, lipid peroxidation, and mtDNA, as a consequence of mitochondrial abnormalities [10][7].
Mitochondria are also a place where ROS are produced in great amounts, mainly as end-products from OXPHOS [122][150]. ROS are usually culprits of mitochondrial damage in cardiovascular diseases, especially when an overproduction of free radicals or impairment of the ROS-scavenging enzyme system occurs [123][124][125][126][151,152,153,154]; moreover, ROS further sustain inflammation [127][155]. In the last few years, evidence involving mtROS in CAVD has become manifold. First studies carried out on isolated ex vivo cultures from human valve samples demonstrated how the presence of lipoprotein a (Lp(a)) selectively triggered acute superoxide production from mitochondria, [128][156] and through it, Lp(a) chronically lead to cell calcification. Lp(a) induces significant calcium deposition in vitro, and patients with CAVD have higher plasma levels of Lp(a) [129][130][157,158]. mtROS reached high levels in a few hours after Lp(a) exposure, but the calcification followed in several days. This evidence ascribes a role for mtROS generation in the acute stages of the disease, but it is still in doubt if they can be targeted to revert the pathological phenotype once the CAVD condition is established.