Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 3 by Rita Xu.
Atherogenic dyslipidemia plays a critical role in the development of metabolic syndrome (MetS), being one of its major components, along with central obesity, insulin resistance, and hypertension.
- atherogenic dyslipidemia
- metabolic syndrome
- cholesterol
1. Introduction
Atherogenic dyslipidemia is defined by increased levels of plasma triglycerides (TG > 150 mg/dL), small dense low-density lipoprotein cholesterol (LDL-C) > 130 mg/dL, total cholesterol (TC) > 200 mg/dL, apolipoprotein B (apoB) and free fatty acids (FFAs) associated with low HDL cholesterol level (HDL-C < 40 mg/dL) and is a major component of metabolic syndrome (MetS) [1]. Metabolic syndrome (MetS) encompasses a group of metabolic disorders that include dyslipidemia, central obesity, hyperglycemia/diabetes mellitus, and hypertension [2][3][4] (Figure 1).
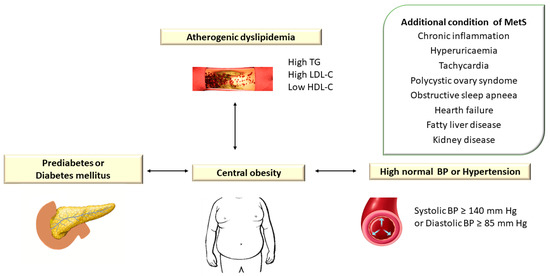
Figure 1. Atherogenic Dyslipidemia Associated with Other Metabolic Disorders in Metabolic Syndrome. TG: triglyceride; LDL-C: low-density lipoprotein cholesterol; HDL-C: high-density lipoprotein cholesterol; BP: blood pressure. In the European guidelines, hypertension is defined as blood pressure (BP) ≥ 140/90 mm Hg, while the American guidelines consider values of TA ≥ 130/80 mm Hg.
The characteristic clinical manifestations of MetS are considered important risk factors for premature cardiovascular diseases (CVD). The prevalence of MetS is estimated between 13 and 36% among the European population [5].
MetS in Europe was 24.3% (23.9% in men vs. 24.6% in women, p < 0.001) in a study by Scuteri et al. [6]. The authors studied 34,821 subjects from 12 cohorts from 10 European countries and 1 of the American participants in the MARE (Metabolic Syndrome and Arteries Research) consortium with the aim of investigating differences in the distribution of “risky” clusters of MetS components according to geographic region and ethnicity. About 12% of subjects with MetS in Europe showed preexisting cluster triglyceride (T)-elevated BP (B)-waist circumference (W) (T-B-W group). This association was more frequent in patients from the UK (32.3%), Italy (19.6%), and Germany (18.5%), compared to Spain (2.6%), Sweden (1.2%), and the USA (2.5%). The G-B-W group was detected in 12.7% of patients with MetS, being more frequent in the population of Southern Europe compared to Northern Europe [6].
The prevalence of MetS (using The National Cholesterol Education Program (NCEP) Adult Treatment Group III (ATP III)) in the United States was 24%, and of the individual components of MetS was 30.0% for hypertriglyceridemia, 37.1% for low levels of HDL cholesterol, 34.0% for hypertension, 38.6% for increased waist circumference (WC), and 12.6% for hyperglycemia [7]. The estimate of the prevalence of MetS in the US National Health and Nutrition Examination Survey (NHANES) 2011–2018 revealed that it increased from 37.6% (95% confidence interval (CI): 34.0–41.4%) in 2011–2012 to 41.8% (95% CI: 38.1–45.7%) in 2017–2018. Among the MetS components, it was estimated that in the case of elevated plasma glucose values, the prevalence increased from 48.9% (95% CI: 45.7–52.5%) in 2011–2012 to 64.7% (95% CI: 61.4–67.9%) in 2017–2018 [8].
The etiopathogenesis of MetS is complex, heterogeneous, and not fully known, being the result of the interaction between genetic and environmental factors [9][10]. In many studies, CVD risk factors are classified into two categories: non-modifiable risk factors (age, sex, family history of CVD) and modifiable risk factors (sedentary lifestyle, smoking, hypercholesterolemia, diabetes mellitus (DM), systolic hypertension) [9][10].
Along with modifiable environmental factors (excess food and sedentary lifestyle), a major role in the etiology of MetS is played by genetic susceptibility (heritability). In recent years, many studies have focused on the study of genetic factors associated with the phenotypic manifestations of MetS. Another metabolic disorder specific to the MetS is insulin resistance. Insulin resistance is associated with increased fasting blood glucose and increases the sensitivity of visceral adipocytes to lipolytic hormones, which causes an increased flow of FFAs to the liver, stimulating hepatic triglyceride and ApoB synthesis [6][11]. Lipoprotein lipase (LPL) mediates the formation of LDL, a major element of dyslipidemia, in muscle tissue and adipose tissue [6][11]. The pathogenic mechanisms involved in the etiology of MetS are complex and most likely the result of the complex interaction between genetic and environmental factors at the level of different types of cellular structures. Over time, numerous candidate genes involved in the regulation of lipid metabolism have been discussed (e.g., polymorphism of the adiponectin, PPARγ, LPL, and CETP genes) [6].
2. The Role of Atherogenic Dyslipidemia in the Metabolic Syndrome
Metabolic syndrome comprises an association of cardiovascular and metabolic risk factors, which include atherogenic dyslipidemia as a major element, along with increased blood pressure and plasma glucose [3][11] (Figure 2).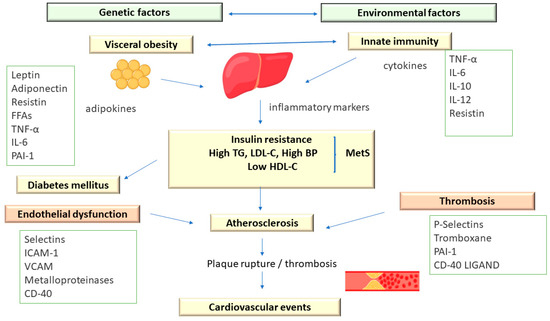
Figure 2. The Pathophysiology of Metabolic Syndrome.
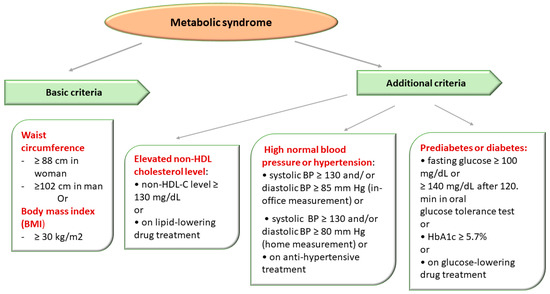
Figure 3. Metabolic Syndrome Diagnostic Criteria Defined in Accordance with The National Cholesterol Education Program Adult Treatment Panel III (NCEP/ATPIII criteria).
3. Heritability of Metabolic Syndrome and Its Components: Current Knowledge
The contribution of genetic factors (heritability) to the occurrence of MetS and its components has been proven by many family-based studies and twin studies [11]. In the study by Carmelli et al. [16], which included 2508 male twin pairs, the concordance for the three MetS components (DM, obesity, and hypertension) was 31.6% for monozygotic twins and 6.3% for dizygotic twins [16]. Lin et al. [14] showed, in a study that included 803 subjects from 89 Caribbean-Hispanic families, that the heritability of MetS (defined in accordance with the National Cholesterol Education Program Adult Treatment Panel III—NCEP/ATPIII criteria) was 24% (p = 0.009), and ranged from 16 to 60% for its five components [14]. The authors demonstrated moderate and significant heritability both for MEtS itself and for its individual components and independent factors that influence MetS [14]. Bellia et al. [17] showed, in the Linosa Study (LiS) group consisting of 293 Caucasian native subjects from 51 families (123 parents; 170 offspring), that the heritability of MetS was 27% (p = 0.0012), and among its individual components, heritability ranged from 10% for blood glucose to 54% for HDL-C. The MetS subtype associated with central obesity, hypertriglyceridemia, and Iow HDL had the highest heritability (31%; p < 0.001) [17]. The marked variability of MetS heritability in different studies could be partly attributed to the race and ethnicity of the individuals included in the study [11][17][18][19]. Starting from the evidence regarding heritability in the case of MetS, but also of its individual components, various studies were carried out that aimed to identify the determining genetic factors both in the case of MetS and its individual components (dyslipidemia, obesity, hypertension, and DM) [11][18][19]. Genetic factors act independently of environmental factors involved both in the etiology of atherogenic dyslipidemia and the other components of MetS, and the variable phenotype is the result of their permanent interaction. The study of monogenic mutations causing atherogenic dyslipidemia as well as polygenic risk factors (genetic polymorphisms and environmental factors) has been carried out by two types of studies: linkage analysis (LA) and association studies (CGS, GWAS, and WES) [20][21]. Linkage analysis (LA) studies investigate the association (co-segregation) between DNA polymorphisms and the onset of the disease and the hereditary inheritance of the disease in the studied family. LA studies are used to identify the involved gene locus and the pathogenic allelic variant, especially in the case of monogenic diseases and less so in the case of multifactorial (polygenic) diseases with complex etiology [21][22]. Association studies using the candidate-gene approach (CGA) represent an alternative method of studying polygenic diseases. CGA analyzes the association between genes and certain diseases, starting from the known pathogenic mechanisms of the disease in which these genes are supposed to intervene. The genes involved in lipid metabolism, obesity, DM, lipoproteins, and hypertension have been analyzed in MetS. Insufficient data on the highly complex pathogenic mechanisms of MetS and its components, however, limit this type of approach. The accelerated development of molecular genetics techniques in recent years has made extensive analyses possible, such as genome-wide association studies (GWASs) and whole exome sequencing (WES), which have provided new information related to the genetic component (heritability) in the case of multifactorial diseases (polygenic) [21][22]. Co-segregation of markers located near candidate genes for MetS and its components helps to identify the loci where those genes (called “positional candidate” genes) are located. The lack of uniform use of the criteria for defining MetS, as well as the multiple combinations between its components (which could be due to several loci specific to a certain association), represent the biggest disadvantage of using LA in the case of MetS. Furthermore, MetS is the consequence of the simultaneous action of multiple genes located on different chromosomes, masking the detection of discrete and unique linkage signals. The studies conducted so far have detected numerous loci involved in the etiology of MetS, located on chromosomes 1q23-31, 3q27, 17p12, chromosome 6 (D6S403, D6S264), and chromosome 7 (D7S479-D7S471) [23][24][25]. In a study that analyzed 2209 individuals with MetS, Kissebach et al. [23] showed that the 3q27 locus was strongly associated with six MetS phenotypic traits, including weight, body mass index (BMI), waist circumference (WC), hip circumference, insulin, and insulin-to-glucose ratio [23]. In the Insulin Resistance Atherosclerosis Family Study (IRAS FS), which included 216 Hispanic individuals with MetS, Langefeld et al. [24] provided evidence for the involvement of the 1q23-q31 locus [24]. In a study that examined patients from North America, Pollex et al. [26] identified several chromosomal regions (1p34.1, 1q41, 2p22.3, 7q31.3, 9p13.1, 9q21.1, 10p11.2, and 19q13.4) related to the occurrence of MetS [26]. A study that included 64 Chinese families revealed a significant association between MetS and its components in the case of some loci located on chromosomes 1, 2, and 16. Furthermore, a region located on chromosome 1q21-q25, known to be linked to T2DM in certain ethnicities, had a significant association with MetS [27]. Other studies that analyzed MetS components individually showed an association between the 5q locus and diastolic blood pressure (DBP); an association between loci 2q, 3q, 6q, 9q, 10q, and 17q and plasma levels of TG; while loci 12p, 12q, and 22q were associated with the plasma level of HDL-C [22][27]. In a study that included 250 German families, Hoffmann et al. [28] suggested that the 1p36.13 locus represents a susceptibility locus for T2DM and MetS [28]. Four other loci located on chromosomes 3p, 3q, 4q, and 14p were associated with T2DM, CVD, and MetS in the Diabetes Heart Study, which included 977 Caucasian subjects [22][29]. The results of these studies demonstrate that none of these loci can be definitively linked to MetS, there being differences at the population level, probably related to ethnicity and race, but also the lack of uniform criteria for defining MetS or false positive results. Taking into account these aspects, the results of LA studies exploring the genetic causes of complex diseases, such as MetS, must be interpreted with caution [22][28][29].References
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188.
- Rezaianzadeh, A.; Namayandeh, S.M.; Sadr, S.M. National Cholesterol Education Program Adult Treatment Panel III Versus International Diabetic Federation Definition of Metabolic Syndrome, Which One is Associated with Diabetes Mellitus and Coronary Artery Disease? Int. J. Prev. Med. 2012, 3, 552–558.
- Dobrowolski, P.; Prejbisz, A.; Kurylowicz, A.; Baska, A.; Burchardt, P.; Chlebus, K.; Dzida, G.; Jankowski, P.; Jaroszewicz, J.; Jaworski, P.; et al. Metabolic syndrome—A new definition and management guidelines: A joint position paper by the Polish Society of Hypertension, Polish Society for the Treatment of Obesity, Polish Lipid Association, Polish Association for Study of Liver, Polish Society of Family Medicine, Polish Society of Lifestyle Medicine, Division of Prevention and Epidemiology Polish Cardiac Society, “Club 30” Polish Cardiac Society, and Division of Metabolic and Bariatric Surgery Society of Polish Surgeons. Arch. Med. Sci. 2022, 18, 1133–1156.
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645.
- Haverinen, E.; Paalanen, L.; Palmieri, L.; Padron-Monedero, A.; Noguer-Zambrano, I.; Sarmiento Suarez, R.; Tolonen, H.; Joint Action on Health Information (InfAct). Comparison of metabolic syndrome prevalence using four different definitions—A population-based study in Finland. Arch. Public Health 2021, 79, 231.
- Scuteri, A.; Laurent, S.; Cucca, F.; Cockcroft, J.; Cunha, P.G.; Manas, L.R.; Mattace Raso, F.U.; Muiesan, M.L.; Ryliskyte, L.; Rietzschel, E.; et al. Metabolic syndrome across Europe: Different clusters of risk factors. Eur. J. Prev. Cardiol. 2015, 22, 486–491.
- Ford, E.S.; Giles, W.H.; Dietz, W.H. Prevalence of the metabolic syndrome among US adults: Findings from the third National Health and Nutrition Examination Survey. JAMA 2002, 287, 356–359.
- Liang, X.; Or, B.; Tsoi, M.F.; Cheung, C.L.; Cheung, B.M.Y. Prevalence of metabolic syndrome in the United States National Health and Nutrition Examination Survey 2011–18. Postgrad. Med. J. 2023, 1–8.
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 2020, 41, 407–477.
- Brown, J.C.; Gerhardt, T.E.; Kwon, E. Risk Factors For Coronary Artery Disease. In StatPearls; StatPearls: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK554410/ (accessed on 29 May 2023).
- Grundy, S.M. Atherogenic dyslipidemia associated with metabolic syndrome and insulin resistance. Clin. Cornerstone 2006, 8, S21–S27.
- Garcia-Giustiniani, D.; Stein, R. Genetics of Dyslipidemia. Arq. Bras. Cardiol. 2016, 106, 434–438.
- Fahed, G.; Aoun, L.; Bou Zerdan, M.; Allam, S.; Bou Zerdan, M.; Bouferraa, Y.; Assi, H.I. Metabolic Syndrome: Updates on Pathophysiology and Management in 2021. Int. J. Mol. Sci. 2022, 23, 786.
- Lin, H.F.; Boden-Albala, B.; Juo, S.H.; Park, N.; Rundek, T.; Sacco, R.L. Heritabilities of the metabolic syndrome and its components in the Northern Manhattan Family Study. Diabetologia 2005, 48, 2006–2012.
- Nesto, R.W. Beyond low-density lipoprotein: Addressing the atherogenic lipid triad in type 2 diabetes mellitus and the metabolic syndrome. Am. J. Cardiovasc. Drugs 2005, 5, 379–387.
- Carmelli, D.; Cardon, L.R.; Fabsitz, R. Clustering of hypertension, diabetes, and obesity in adult male twins: Same genes or same environments? Am. J. Hum. Genet. 1994, 55, 566–573.
- Bellia, A.; Giardina, E.; Lauro, D.; Tesauro, M.; Di Fede, G.; Cusumano, G.; Federici, M.; Rini, G.B.; Novelli, G.; Lauro, R.; et al. “The Linosa Study”: Epidemiological and heritability data of the metabolic syndrome in a Caucasian genetic isolate. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 455–461.
- Stancakova, A.; Laakso, M. Genetics of metabolic syndrome. Rev. Endocr. Metab. Disord. 2014, 15, 243–252.
- Khan, R.J.; Gebreab, S.Y.; Sims, M.; Riestra, P.; Xu, R.; Davis, S.K. Prevalence, associated factors and heritabilities of metabolic syndrome and its individual components in African Americans: The Jackson Heart Study. BMJ Open. 2015, 5, e008675.
- Jo, G.; Kwak, S.Y.; Kim, J.Y.; Lim, H.; Shin, M.J. Association between Genetic Variant of Apolipoprotein C3 and Incident Hypertension Stratified by Obesity and Physical Activity in Korea. Nutrients 2018, 10, 1595.
- Abou Ziki, M.D.; Mani, A. Metabolic syndrome: Genetic insights into disease pathogenesis. Curr. Opin. Lipidol. 2016, 27, 162–171.
- Joy, T.; Lahiry, P.; Pollex, R.L.; Hegele, R.A. Genetics of metabolic syndrome. Curr. Diab. Rep. 2008, 8, 141–148.
- Kissebah, A.H.; Sonnenberg, G.E.; Myklebust, J.; Goldstein, M.; Broman, K.; James, R.G.; Marks, J.A.; Krakower, G.R.; Jacob, H.J.; Weber, J.; et al. Quantitative trait loci on chromosomes 3 and 17 influence phenotypes of the metabolic syndrome. Proc. Natl. Acad. Sci. USA 2000, 97, 14478–14483.
- Langefeld, C.D.; Wagenknecht, L.E.; Rotter, J.I.; Williams, A.H.; Hokanson, J.E.; Saad, M.F.; Bowden, D.W.; Haffner, S.; Norris, J.M.; Rich, S.S.; et al. Linkage of the metabolic syndrome to 1q23-q31 in Hispanic families: The Insulin Resistance Atherosclerosis Study Family Study. Diabetes 2004, 53, 1170–1174.
- Arya, R.; Blangero, J.; Williams, K.; Almasy, L.; Dyer, T.D.; Leach, R.J.; O’Connell, P.; Stern, M.P.; Duggirala, R. Factors of insulin resistance syndrome—Related phenotypes are linked to genetic locations on chromosomes 6 and 7 in nondiabetic mexican-americans. Diabetes 2002, 51, 841–847.
- Pollex, R.L.; Hegele, R.A. Genetic determinants of the metabolic syndrome. Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 482–489.
- Ng, M.C.; So, W.Y.; Cox, N.J.; Lam, V.K.; Cockram, C.S.; Critchley, J.A.; Bell, G.I.; Chan, J.C. Genome-wide scan for type 2 diabetes loci in Hong Kong Chinese and confirmation of a susceptibility locus on chromosome 1q21-q25. Diabetes 2004, 53, 1609–1613.
- Hoffmann, K.; Mattheisen, M.; Dahm, S.; Nurnberg, P.; Roe, C.; Johnson, J.; Cox, N.J.; Wichmann, H.E.; Wienker, T.F.; Schulze, J.; et al. A German genome-wide linkage scan for type 2 diabetes supports the existence of a metabolic syndrome locus on chromosome 1p36.13 and a type 2 diabetes locus on chromosome 16p12.2. Diabetologia 2007, 50, 1418–1422.
- Bowden, D.W.; Rudock, M.; Ziegler, J.; Lehtinen, A.B.; Xu, J.; Wagenknecht, L.E.; Herrington, D.; Rich, S.S.; Freedman, B.I.; Carr, J.J.; et al. Coincident linkage of type 2 diabetes, metabolic syndrome, and measures of cardiovascular disease in a genome scan of the diabetes heart study. Diabetes 2006, 55, 1985–1994.
More