Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Kostis Gyftopoulos and Version 2 by Jessie Wu.
Homologous recombination deficiency (HRD) is a term describing tumor phenotypes in which the ability to repair DNA double-strand breaks utilizing the homologous recombination repair (HRR) pathway is lost. Lately, precision medicine has been focusing on targetable mutations, although their frequency in tumors may be very low. The most notable mutations that can be targeted in PCa include gene products that regulate DNA repair through homologous recombination (HR), such as BRCA1, BRCA2, ATM, PALB2, CHEK2 and HOXB13.
- prostate cancer
- DNA damage repair
- homologous recombination deficiency
- biomarkers
1. Introduction
Homologous recombination repair (HRR) is part of the DNA damage repair (DDR) pathway that also includes base excision repair, nucleotide excision repair, mismatch repair and non-homologous end joining [1][2]. HRR is activated during the S and G2 phases of the cell cycle and is a very efficient and error-free process, in contrast to non-homologous end joining, which repairs errors at any phase of the cell cycle and has a propensity for errors [1]. The importance of such mechanisms is substantial, as it defines the fate of the cell after DNA damage [3]. After exposure to carcinogens, including endogenous or exogenous factors such as ultraviolet and ionizing radiation and chemical pollutants that cause oxidative stress, DNA damage repair (DDR)R is activated before the cell commences replication [3][4]. DDR recruits tumor suppressor proteins and restrains the aggregation of genomic alterations [2]. If the damage is extensive, then cell death occurs, since the accumulation of double- and single-strand breaks may lead to genomic instability, which carries a risk of malignant transformation [3][4][5].
DNA repair enzymes are found mutated in a variable proportion of prostate cancer (PCa) cases, ranging from 5–10% in localized disease to almost 20% in advanced, castrate-resistant or metastatic disease [1][2][3][6][7][8][9][10]. Analysis of 333 cases of primary PCa retrieved from The Cancer Genome Atlas (TCGA) demonstrated that mutations in HR genes were detected in 19% of the samples, being present even at the early stages of the disease [4][11]. The most common alterations in the TCGA database were ATM mutations (4%), RAD51C deletions (3%), BRCA2 deletions or mutations (3%), CKD12 deletions or mutations (2%), BRCA1 mutations (1%) and FANCD2 aberrations (6%) [4]. In a study including 150 patients with recurrent or metastatic PCa, without any further selection, 14% of the patients were found to harbor pathogenic mutations for enzymes that mediated DNA damage repair in their tumors and BRCA2 was the most commonly affected gene [10][12]. HRD gene aberrations are considered an early-stage event in PCa, but their more frequent occurrence in advanced disease is explained by their association with a poor prognosis [13] and their role in disease progression [2].
2. Germline vs. Sporadic Mutations in Homologous Recombination Genes
Alterations in HR genes are most commonly sporadic but can also be germline [14]. The term inherited PCa refers to families that fulfill the John Hopkins criteria, these being the following: (a) at least three first-degree relatives diagnosed with PCa, (b) the presence of the disease in three consecutive generations and (c) early-onset disease in two family members [15].
It is well established that a family history of PCa increases the risk for all male relatives, even in the absence of characteristic genetic alterations [16][17]. These cases, comprising 15–20% of all patients, are best described with the term “family-associated PCa” and should not be confused with hereditary PCa, which occurs less frequently, at an estimated 5% [17], and includes a population with a specific molecular profile—for instance, BRCA1/2 or HOXB13 mutation carriers [16]. A proposed theory is that the predisposition in families with a higher incidence of PCa probably occurs due to the interplay between common polymorphisms of intermediate and low penetrance in various genes with environmental risk factors that enhance inflammation [17].
A hereditary predisposition to PCa should be suspected when there is a family member diagnosed with PCa at an age < 60 years or with an aggressive disease course, a family history of more than three malignancies related to hereditary breast/ovarian cancer or Lynch syndrome or, finally, in men of Ashkenazi Jewish origin [18] and if two distinct histological patterns are seen in a prostate biopsy: intraductal carcinoma of the prostate (IDCp) and cribriform histology (see below) [12][16][19][20].
The PRACTICAL study analyzed germline mutations in men predisposed to PCa and demonstrated that mutations that are considered pathogenic or likely pathogenic in genome databases (for instance, ClinVar) were linked with a worse prognosis, a fact that was not observed for mutations classified as variants of uncertain significance. This means that the detection of a mutation or variant alone is not informative, and further characterization of the genetic alteration detected is required in order to obtain accurate predictive information for the patient [21].
Germline mutations in DDR have distinct behavior and malignant potential compared with sporadic cases [4]. Somatic mutations can develop after progression to metastatic CRPC (mCRPC) [8], while germline mutations are inherited through an autosomal dominant pathway with incomplete penetrance [1]. Germline BRCA2 mutations have especially been associated with a poor prognosis and have been found to be an independent prognostic factor for PCa patients [22][23], and this applies even in cases with a limited tumor volume and low histopathological grade [8]. These neoplasms demonstrate higher genomic instability and more copy number alterations [8], including MYC amplification, which is known to correlate with aggressive behavior and rapid disease progression [24]. The aggressiveness of BRCA2-mutated neoplasms has been attributed to the fact that these tumors develop a subpopulation of cells that are castrate-resistant and can grow independently, even after the administration of antiandrogen therapy. This model is supported by research data that show a similar molecular signature in BRCA2-mutated tumors and metastatic CRPC, which is only rarely found in sporadic PCa [8]. Another theory is that these neoplasms, due to the DNA repair defects that they harbor, gradually accumulate genetic alterations, in contrast to sporadic cases with functionable DNA repair systems, where DNA defects are properly and timely repaired [8]. It has been proposed that genetic alterations, characterized as truncal, arise at the early stages of carcinogenesis and are carried by all the daughter cells, while later-acquired alterations are present only in specific cell subpopulations, contributing to the heterogeneity and complexity of cancer genetics [16].
The presence of germline mutations has additional implications for the relatives of the patient, as they should be tested as potential carriers [6][7][16]. Of interest, 5.5% of men with a familial predisposition to PCa share the same mutational pattern in DNA repair enzymes, such as BRCA1, BRCA2 and ATM alterations, even if they have not developed PCa [6]. Experts suggest to start screening for PCa in men with known BRCA2 and BRCA1 mutations at the age of 40 [7], supported by studies that have revealed a diagnosis of PCa at as early as 41 years of age in BRCA2- and 43 in BRCA1-mutated patients [25].
3. BRCA and Non-BRCA Mutations
BRCA mutations represent the most common DNA repair alterations in PCa [1] and, among them, the majority of cases show BRCA2 alterations (12% BRCA2 alterations versus 2% BRCA1 in advanced PCa) [2][4][13][16]. The most common BRCA2 alteration results in the production of a truncated form of the protein, followed by complete deletion of the gene; only a minority of cases show point mutations [16]. In contrast, the most frequent BRCA1 mutations lead to a truncated gene product and are often accompanied by TP53 mutations [26].
HOXB13 was the first gene that was shown to enhance the prostate cancer risk by up to 10 times and was linked with familial cases of PCa and early-onset disease in some [7][15][17] but not all studies [27]. Specifically, the mutant HOXB13G84E has been associated with lower-risk tumor characteristics [27], early-onset disease [17] and European origin among patients [15][17]. Other alterations have been encountered in different populations, such as G135E in Chinese men and variants A128D and F240L in Portuguese men [15].
A recent study conducting genome analysis on non-BRCA-mutated PCa showed that germline ATM and CHEK2 alterations had lower penetrance than BRCA2. In addition, prostate cancer carried different genetic alterations in these genes compared to breast and ovarian cancer [27]. Germline CHEK2 mutations increase the risk for PCa development at a moderate level [7] and have been linked with aggressive or high-risk cancer [27]. Genome analysis in patients with non-BRCA-mutated familial PCa has confirmed that mutated ATM is found in cases with advanced disease, higher PSA levels at the time of initial diagnosis and a high D’Amico score [27]. Further studies need to be performed, mainly for ATM and PALB2, as the existing data for these two genes in PCa are limited. It is promising, though, that ATM aberration augments the sensitivity to PARP inhibitors [28].
CDK12 is a cyclin-dependent kinase that regulates transcription elongation through the phosphorylation of RNA polymerase II, subsequently modifying gene expression and influencing DDR gene expression [29]. CDK12 is mutated in a small percentage of metastatic castrate-resistant PCa (CRPC), varying from 4.7% to 7%, and, when mutated, it is not combined with HR deficiency and ATM or MMR gene mutations [30][31]. Thus, CDK12-mutant PCa comprises a distinct molecular group of PCa.
4. Clinical and Histologic Characteristics of Homologous Recombination DeficiencyD Tumors
BRCA2-mutated tumors tend to develop in younger patients, are usually classified as an intermediate or high risk of recurrence, metastasize earlier and are associated with shorter survival, even when treated with prostatectomy or radiotherapy [2][7][8][11][20][25][32][33]. In addition, HRD-targeted therapies have been developed (see below). Thus, tumor testing for HR genes is recommended in all metastatic PCa patients and can be considered in patients with regional disease, especially those with adverse characteristics [34].
The IMPACT study focused on screening men having BRCA1 and BRCA2 mutations for the diagnosis of PCa and revealed that the value of prostate-specific antigen (PSA) levels higher than 3.0 ng/mL and of prostate biopsy was greater in the BRCA2-mutated population than in the BRCA2 wild type [25]. In contrast to these data, there are increasing data available showing that PCa with low PSA values at the time of diagnosis is associated with DDR mutations [12], and, in comparison, metastatic cases with a known BRCA2 [35] but not BRCA1 [25] mutation present with lower PSA levels compared to their wild-type controls. Another study that tried to elucidate the pathological characteristics of BRCA2-mutated tumors exhibited no statistically significant difference compared to BRCA2 wild-type tumors regarding the TNM classification, prognostic grade group or histology subtype of the tumors [35]. The mutated subgroup, however, had a higher mutational load and recurring ATM and BRCA1 alterations [35].
Intraductal carcinoma, which is associated with high-grade and high-stage PCa; the presence of lymph node and distant metastases; and shorter disease-specific and overall survival is more frequently seen in cases of hereditary PCa and often harbors BRCA1/2 mutations [20][36][37][38][39][40]. Additionally, BRCA2 mutation carriers have a higher probability of showing IDCp in their biopsy [8][20][36][41][42][43][44]. Even IDCp associated with low-grade PCa has been shown to harbor aberrations in DDR genes, such as BRCA2, CHEK2 and CDK12, which are not present in the invasive component [45]. Furthermore, according to recent data, in PCa without IDCp, HRD (estimated by a higher HRD score—see below) results from mutations in DDR genes, in contrast to PCa with IDCp, where HRD is attributed to TP53 mutations [46]. Whole-genome sequencing of BRCA2-mutated and IDCp-harboring PCa revealed the molecular resemblance of these tumors to metastatic CRPC, even at the initial stages of tumorigenesis, and the activation of crucial signaling pathways, such as WNT/b-catenin modulator MED12L/MED12, which have been associated with an adverse prognosis [20][47]. It should be mentioned that these alterations are not found in sporadic cancers with IDCp, while MED12 is absent in normal prostate and organ-confined PCa [20].
Apart from intraductal carcinoma, the somatic loss of both alleles of the BRCA2 gene and increased genomic instability and copy number alterations have also been associated with the cribriform pattern and the ductal type of adenocarcinoma [19][44][47][48].
Based on these data, current guidelines suggest that patients with intermediate-risk PCa and IDCp or cribriform histology can be considered for germline or somatic genetic testing for DDR alterations [49][50][51][52][53][54][55][56].
5. Clinical and Therapeutic Implications of Homologous Recombination Deficiency
Based on the aggressiveness of HRD-mutated neoplasms, an earlier and more aggressive therapeutic approach should be followed for BRCA2-mutated tumors. Moreover, patients with this molecular signature demonstrate a significant response to platinum-based chemotherapy and poly-adenosine diphosphate (ADP) ribose polymerase (PARP) inhibitors (PARPi) (see below). Regarding platinum-based chemotherapy, HRD has been associated with an increased likelihood of a PSA response in a small cohort (N = 64) of patients with PCa, although no difference in overall survival was seen [57]. However, the number of patients enrolled in this cohort was limited, so these observations need to be validated in larger groups of patients [57][58]. Similarly, a small prospective cohort study showed that patients with germline mutations in DDR experienced better outcomes when treated with abiraterone or enzalutamide, compared to taxanes [22]. In addition, radical prostatectomy, rather than radiotherapy, should be the treatment of choice for these patients in the localized setting [8]. Interestingly, these worrisome features are not detected in BRCA1 carriers, indicating that the clinical implications of these two mutations are significantly different [25].
Patients with CRPC and HRD alterations show promising response rates after treatment with PARP inhibitors [28]. Therefore, in 2016, PARP inhibitors received approval by the Food and Drug Administration (FDA) and were incorporated into the therapeutic schemes of metastatic CRPC [28]. To date, two PARP inhibitors (Olaparib and Rucaparib) have been approved for metastatic CRPC [59][60]. Clinical data support their efficacy, as documented by PSA and circulating tumor cell responses and improved progression-free survival and overall survival [60].
One of the first clinical trials that elucidated their utility was the TOPAPR-A trial [61]. The patient group included fifty (50) patients harboring alterations in DNA repair enzymes, previously treated with docetaxel or second-generation androgen deprivation therapy (ADT); treatment with Olaparib resulted in a favorable clinical response [61]. Based on the subsequent PROfound clinical trial (NCT02987543) [62] (a randomized phase 3 clinical trial in 245 patients with a mutation in at least one of BRCA1, BRCA2 and ATM and 152 patients with alterations in other HRD genes), Olaparib was approved for patients whose tumors harbor a genetic alteration in BRCA1, BRCA2, ATM, BRIP1, BARD1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D or RAD54L as a second-line therapy after the failure of second-generation antiandrogen agents or docetaxel or as a third-line therapy [7]. Based on the Triton 2 (NCT02952534) and Triton 3 (NCT02975934) clinical trials [63][64], Rucaparib has been approved for tumors harboring BRCA1/2 mutations, either somatic or germline [65][66]. Currently, ongoing clinical trials are tested the efficiency of other members of the PARP inhibitor family, such as niraparib (clinical trial number: NCT02854436) [7].
Proper risk assessment of patients at the time of the initial diagnosis should incorporate the HRD status [46]. Interestingly, different alterations in the genes have recently been found to result in different response rates to treatment [67]. For instance, a PCa patient with a base substitution (c.4211C > G) in BRCA2 showed a response to radiotherapy and androgen deprivation therapy (ADT) in a Chinese cohort study [68], while patients with CDK12 mutations did not respond well to hormonal therapy, PARPi or taxanes but showed positive (and occasionally durable) responses to PD-1 inhibition [69][70]. On the contrary, ATM and CDK12 mutations do not seem to respond to PARP inhibitors as effectively as BRCA1/2 mutations [71][72]. Despite the fact that this observation was noticed in a small group of patients (46 patients) with progressive metastatic CRPC, and the retrospective nature of the study, it is in accordance with the results of the Triton 3 trial [71]. A possible reason for this difference is that biallelic loss and germline mutations, which are usually detected in BRCA1/2 carriers, respond better to the treatment [71]. This underlines the importance of accurate sequencing in HR genes in order to be fully utilized as both prognostic and predictive biomarkers.
6. Predictive Biomarkers to Poly-Adenosine Diphosphate Ribose Polymerase InhibitorsRPi Response
Next-generation sequencing (NGS) seems to be the most appropriate tool to detect alterations in the HRD-associated genes mentioned above. This method detects multiple genetic alterations, including mutations and chromosomal alterations in a single test, although none of the currently available tests is validated to detect germline mutations. Genomic analysis with a high reading depth can raise the awareness of hereditary PCa, and these cases should be referred to genetic counseling that provides an holistic approach and guides the patients through specialized genetic tests [16]. NGS testing can be performed on metastatic tissue or on plasma circulating free DNA (cfDNA) [16]. Patients with mutations in genes other than BRCA1/2, such as ATM, PALB2, CHEK2, FANCA and HDAC2, are also responding well to PARP inhibitors, underlying the importance of using a broader detection panel [28].
A scoring system, called the Homologous Recombination Deficiency Score, has also been established, incorporating several chromosomal aberrations, such as the loss of heterozygosity, telomeric allelic imbalance and large-scale transitions [46]. This score gives, however, a general expression of the HRD status and does not directly reflect which particular enzyme is damaged. Nonetheless, it appears that it can be successfully utilized as a predictive biomarker for the potential response to PARP inhibitors [46]. The presence of MYC and TP53 alterations is also frequently associated with high HRD scores, even without synchronous aberrations in the HR system [46]. The MYC oncogene supervises the repair of double-strand DNA breaks [46]. Subsequently, the concurrent inhibition of the MYC pathway along with PARP inhibitors could be beneficial [46].
Regarding the follow-up of patients under PARP inhibitor treatment, a relatively new but promising approach is the whole-exome sequencing of liquid biopsy specimens, which reduces the need for additional surgical interventions in patients [11][58][73][74]. Testing of metastatic tissue poses some practical difficulties, as metastatic foci in PCa are mostly found in the bones, which sometimes are difficult to access; even when the sample is adequate, the DNA that is extracted from this tissue has questionable quality, due to the decalcification that is performed during tissue processing [75].
One of the first applications of liquid biopsy in PCa research was conducted in the TOPAPR-A clinical trial, where it was depicted that cfDNA analysis can provide adequate information regarding acquired genetic alterations, even before signs of clinical progression are evident, allowing the early detection of resistance [2]. The broad utility of this approach may lead to modifications of the therapeutic scheme and the discontinuation of non-responsive drugs, avoiding unnecessary toxicity [73]. cfDNA is derived from the circulated tumor DNA and DNA fragments that are produced after cellular death or apoptosis [74]. The main disadvantage of this revolutionary method is the small number of circulating tumor cells in some cases, and, thus, the practical difficulty to isolate and further process them in order to extract DNA [58]. Therefore, liquid biopsy is preferably utilized in advanced PCa cases and not in the early stages of the disease.
7. Poly-Adenosine Diphosphate Ribose Polymerase InhibitorsRPi Mechanism of Action
The mechanism of action of PARP inhibitors in HRD tumors has been clarified during the last decade [76]. Normally, the PARP complex consists of 16 enzymes and their common feature is the production of poly(ADP-ribose) from NAD, a chemical reaction that generates nicotinamide [76]. Some members of the PARP family, PARP1 and -2, activate the repair mechanisms after DNA damage [59]. In particular, PARP1 can restore double- and single-strand breaks in the nucleotide chain [59], preserving the integrity of the replication fork and, subsequently, of transcription, thus shielding the genome against replication stress [76]. If this process fails, then replication is interrupted, and deadly breaks, followed by cellular death, are induced [76]. Homologous recombination undertakes the correction of double-strand breaks by enlisting various repair enzymes, including BRCA1 and BRAC2 [76]. PARP1 has a central role in the recruitment of other family members, as the absence of PARP1 downgrades the efficacy of PARP inhibitors in general [76] (Figure 1). Furthermore, in experimental models, PARP1 enhances the oncogenic actions of TMPRSS-ERG, through the enrichment of AR-mediated transcription, which eventually drives the cells into a castrate-resistant phase [2]. PARP1 is essential for the activation of ERG [2].
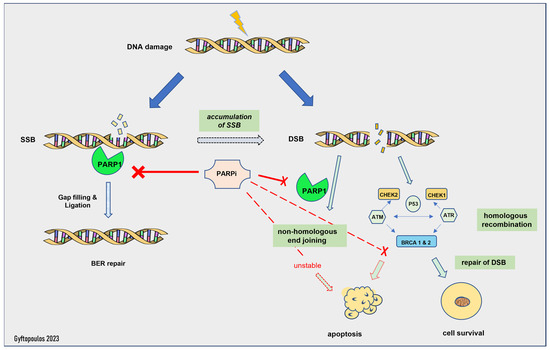
Figure 1.
Mechanism of action of PARP inhibitors. SSB: single-strand break, DSB: double-strand break, BER: base excision repair.
The activity of PARP inhibitors is described as PARP trapping, as it traps PARP1/2 near the region of DNA damage, resulting in the stalling of replication forks. Stalled replication forks lead to highly cytotoxic double-strand breaks that, in HR-proficient cells, are repaired by HR [3][60]. HRD cells are unable to repair the accumulating double-strand breaks and die [6]. Thus, PARP1i is effective only in HRD cells, as the HR-proficient cells can escape its action. This is called the synthetic lethality hypothesis [77]. Synthetic lethality describes a situation where a combination of two events leads to cell death, but each event is individually viable. Olaparib inhibits PARP1 and -2, while Rucaparib is a less selective PARP inhibitor with a broader range of action, including non-PARP targets [76]. Of note, patients with germline mutations in the BRCA genes suffer from severe adverse effects and especially myelotoxicity [3], whereas most patients with sporadic HRD face milder toxicity, such as anemia and fatigue [61].
Therapeutic synthetic lethality can also be applied to tumors that have a molecular signature similar to BRCA-mutated tumors, even in the absence of homologous recombination deficiency, introducing a new term of “BRCAness”. For example, PARP inhibitors have been shown to induce replication stress in experimental models that exhibit the concurrent loss of P53 and RB1 and MYC amplification [1][76]. This could explain the favorable response to these drugs, even in the absence of BRCA mutations [28]. However, despite this experimental evidence, the MAGNITUDE trial failed to show a survival benefit in men who did not harbor HRR mutations and were treated with PARPi (niraparib) combined with abiraterone [72]. On the other hand, platinum-based chemotherapy, such as docetaxel and cabazitaxel, acts through DNA alkylation, producing DNA strand breaks, thus contributing to synthetic lethality. Therefore, they are widely used in advanced PCa [16], although they do not directly target a specific DNA repair mechanism [2].
8. Biomarkers Predicting Poly-Adenosine Diphosphate Ribose Polymerase InhibitorsARPi Resistance
Unfortunately, neoplastic cells eventually develop resistance mechanisms that overcome the external PARP inhibition and block the pathway of synthetic lethality, as the PARP enzymes become functionable again [1]. The time frame in which resistance develops is usually after 10–18 months of treatment [2]. It usually happens through the mutational reversion of BRCA1/2, most frequently due to single-nucleotide alterations that provoke frame shift modifications and result in HR proficiency, preventing the deaths of neoplastic cells [1]. Alternatively, they protect the replication fork to preserve transcription. Other possible resistance mechanisms include the acquisition of genetic alterations in PARP enzymes or the development of efflux pumps that reduce the concentrations of PARP inhibitors within the cancer cells [1]. In a published case report, acquired resistance due to AKT mutation appeared a few months after Olaparib administration and was handled with a concurrent AKT inhibitor [78].
Recent research work proposes that the MMS22L gene (which encodes the DNA repair methyl methanesulfonate-sensitivity 22-like protein) is frequently deleted in PCa, mediates HRR and has predictive value regarding PARP inhibitors’ effectiveness. The suggested mechanism involves the blockage of the RAD51 molecule, an essential moderator of HRR, in a TP53-dependent way [60]. In contrast, the loss of CHEK2 has been found to increase the resistance to PARP inhibitors, due to the upregulation of BRCA2, and the concomitant use of PARP and ATR inhibitors could overcome this resistance pathway [60].
References
- Shah, S.; Rachmat, R.; Enyioma, S.; Ghose, A.; Revythis, A.; Boussios, S. BRCA Mutations in Prostate Cancer: Assessment, Implications and Treatment Considerations. Int. J. Mol. Sci. 2021, 22, 12628.
- Kornberg, Z.; Chou, J.; Feng, F.Y.; Ryan, C.J. Prostate cancer in the era of “Omic” medicine: Recognizing the importance of DNA damage repair pathways. Ann. Transl. Med. 2018, 6, 161.
- Wang, M.; Chen, S.; Ao, D. Targeting DNA repair pathway in cancer: Mechanisms and clinical application. Medcomm 2021, 2, 654–691.
- Schiewer, M.J.; Knudsen, K.E. DNA Damage Response in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2019, 9, a030486.
- Huang, R.-X.; Zhou, P.-K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60.
- Sekhoacha, M.; Riet, K.; Motloung, P.; Gumenku, L.; Adegoke, A.; Mashele, S. Prostate Cancer Review: Genetics, Diagnosis, Treatment Options, and Alternative Approaches. Molecules 2022, 27, 5730.
- Giri, V.N.; Morgan, T.M.; Morris, D.S.; Berchuck, J.E.; Hyatt, C.; Taplin, M. Genetic testing in prostate cancer management: Considerations informing primary care. CA. Cancer J. Clin. 2022, 72, 360–371.
- Taylor, R.A.; Fraser, M.; Rebello, R.J.; Boutros, P.C.; Murphy, D.G.; Bristow, R.G.; Risbridger, G.P. The influence of BRCA2 mutation on localized prostate cancer. Nat. Rev. Urol. 2019, 16, 281–290.
- Miyahira, A.K.; Sharp, A.; Ellis, L.; Jones, J.; Kaochar, S.; Larman, H.B.; Quigley, D.A.; Ye, H.; Simons, J.W.; Pienta, K.J.; et al. Prostate cancer research: The next generation; report from the 2019 Coffey-Holden Prostate Cancer Academy Meeting. Prostate 2020, 80, 113–132.
- Mollica, V.; Marchetti, A.; Rosellini, M.; Nuvola, G.; Rizzo, A.; Santoni, M.; Cimadamore, A.; Montironi, R.; Massari, F. An Insight on Novel Molecular Pathways in Metastatic Prostate Cancer: A Focus on DDR, MSI and AKT. Int. J. Mol. Sci. 2021, 22, 13519.
- Lozano, R.; Castro, E.; Aragón, I.M.; Cendón, Y.; Cattrini, C.; López-Casas, P.P.; Olmos, D. Genetic aberrations in DNA repair pathways: A cornerstone of precision oncology in prostate cancer. Br. J. Cancer 2021, 124, 552–563.
- Velho, P.I.; Silberstein, J.L.; Markowski, M.C.; Luo, J.; Lotan, T.L.; Isaacs, W.B.; Antonarakis, E.S. Intraductal/ductal histology and lymphovascular invasion are associated with germline DNA-repair gene mutations in prostate cancer. Prostate 2018, 78, 401–407.
- Malik, A.; Srinivasan, S.; Batra, J. A New Era of Prostate Cancer Precision Medicine. Front. Oncol. 2019, 9, 1263.
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228.
- Brandão, A.; Paulo, P.; Teixeira, M.R. Hereditary Predisposition to Prostate Cancer: From Genetics to Clinical Implications. Int. J. Mol. Sci. 2020, 21, 5036.
- McNevin, C.S.; Cadoo, K.; Baird, A.-M.; Murchan, P.; Sheils, O.; McDermott, R.; Finn, S. Pathogenic BRCA Variants as Biomarkers for Risk in Prostate Cancer. Cancers 2021, 13, 5697.
- Tan, S.-H.; Petrovics, G.; Srivastava, S. Prostate Cancer Genomics: Recent Advances and the Prevailing Underrepresentation from Racial and Ethnic Minorities. Int. J. Mol. Sci. 2018, 19, 1255.
- Schaeffer, E.; Srinivas, S.; Antonarakis, E.S.; Armstrong, A.J.; Bekelman, J.E.; Cheng, H.; D’Amico, A.V.; Davis, B.J.; Desai, N.; Dorff, T.; et al. NCCN Guidelines Insights: Prostate Cancer, Version 1.2021. J. Natl. Compr. Cancer Netw. 2021, 19, 134–143.
- Hesterberg, A.B.; Gordetsky, J.B.; Hurley, P.J. Cribriform Prostate Cancer: Clinical Pathologic and Molecular Considerations. Urology 2021, 155, 47–54.
- Taylor, R.A.; Fraser, M.; Livingstone, J.; Espiritu, S.M.G.; Thorne, H.; Huang, V.; Lo, W.; Shiah, Y.-J.; Yamaguchi, T.N.; Sliwinski, A.; et al. Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nat. Commun. 2017, 8, 13671.
- Karlsson, Q.; Brook, M.N.; Dadaev, T.; Wakerell, S.; Saunders, E.J.; Muir, K.; Neal, D.E.; Giles, G.G.; MacInnis, R.J.; Thibodeau, S.N.; et al. Rare Germline Variants in ATM Predispose to Prostate Cancer: A PRACTICAL Consortium Study. Eur. Urol. Oncol. 2021, 4, 570–579.
- Castro, E.; Romero-Laorden, N.; del Pozo, A.; Lozano, R.; Medina, A.; Puente, J.; Piulats, J.M.; Lorente, D.; Saez, M.I.; Morales-Barrera, R.; et al. PROREPAIR-B: A Prospective Cohort Study of the Impact of Germline DNA Repair Mutations on the Outcomes of Patients With Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2019, 37, 490–503.
- Castro, E.; Goh, C.; Leongamornlert, D.; Saunders, E.; Tymrakiewicz, M.; Dadaev, T.; Govindasami, K.; Guy, M.; Ellis, S.; Frost, D.; et al. Effect of BRCA Mutations on Metastatic Relapse and Cause-specific Survival After Radical Treatment for Localised Prostate Cancer. Eur. Urol. 2015, 68, 186–193.
- Qiu, X.; Boufaied, N.; Hallal, T.; Feit, A.; de Polo, A.; Luoma, A.M.; Alahmadi, W.; Larocque, J.; Zadra, G.; Xie, Y.; et al. MYC drives aggressive prostate cancer by disrupting transcriptional pause release at androgen receptor targets. Nat. Commun. 2022, 13, 2559.
- Page, E.C.; Bancroft, E.K.; Brook, M.N.; Assel, M.; Al Battat, M.H.; Thomas, S.; Taylor, N.; Chamberlain, A.; Pope, J.; Raghallaigh, H.N.; et al. Interim Results from the IMPACT Study: Evidence for Prostate-specific Antigen Screening in BRCA2 Mutation Carriers. Eur. Urol. 2019, 76, 831–842.
- Dall’Era, M.A.; McPherson, J.D.; Gao, A.C.; DeVere White, R.W.; Gregg, J.P.; Lara, P.N. Germline and somatic DNA repair gene alterations in prostate cancer. Cancer 2020, 126, 2980–2985.
- Mondschein, R.; Bolton, D.; Clouston, D.; Dowty, J.; Kavanagh, L.; Murphy, D.; Scott, P.; Taylor, R.A.; Thorne, H. Novel Germline Mutations in a Cohort of Men with Familial Prostate Cancer. Cancers 2022, 14, 3623.
- Helleday, T. PARP inhibitor receives FDA breakthrough therapy designation in castration resistant prostate cancer: Beyond germline BRCA mutations. Ann. Oncol. 2016, 27, 755–757.
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10, 1757.
- Wu, Y.-M.; Cieślik, M.; Lonigro, R.J.; Vats, P.; Reimers, M.A.; Cao, X.; Ning, Y.; Wang, L.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 2018, 173, 1770–1782.e14.
- Udager, A.M.; Tomlins, S.A. Molecular Biomarkers in the Clinical Management of Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030601.
- Cheng, H.H.; Sokolova, A.O.; Schaeffer, E.M.; Small, E.J.; Higano, C.S. Germline and Somatic Mutations in Prostate Cancer for the Clinician. J. Natl. Compr. Cancer Netw. 2019, 17, 515–521.
- Han, H.; Park, C.K.; Cho, N.H.; Lee, J.; Jang, W.S.; Ham, W.S.; Choi, Y.D.; Cho, K.S. Characteristics of BRCA2 Mutated Prostate Cancer at Presentation. Int. J. Mol. Sci. 2022, 23, 13426.
- Pantazopoulos, H.; Diop, M.-K.; Grosset, A.-A.; Rouleau-Gagné, F.; Al-Saleh, A.; Boblea, T.; Trudel, D. Intraductal Carcinoma of the Prostate as a Cause of Prostate Cancer Metastasis: A Molecular Portrait. Cancers 2022, 14, 820.
- Robinson, B.; Magi-Galluzzi, C.; Zhou, M. Intraductal carcinoma of the prostate. Arch. Pathol. Lab. Med. 2012, 136, 418–425.
- Efstathiou, E.; Abrahams, N.A.; Tibbs, R.F.; Wang, X.; Pettaway, C.A.; Pisters, L.L.; Mathew, P.F.; Do, K.-A.; Logothetis, C.J.; Troncoso, P. Morphologic characterization of preoperatively treated prostate cancer: Toward a post-therapy histologic classification. Eur. Urol. 2010, 57, 1030–1038.
- Kimura, K.; Tsuzuki, T.; Kato, M.; Saito, A.M.; Sassa, N.; Ishida, R.; Hirabayashi, H.; Yoshino, Y.; Hattori, R.; Gotoh, M. Prognostic value of intraductal carcinoma of the prostate in radical prostatectomy specimens. Prostate 2014, 74, 680–687.
- Van Der Kwast, T.; Al Daoud, N.; Collette, L.; Sykes, J.; Thoms, J.; Milosevic, M.; Bristow, R.G.; Van Tienhoven, G.; Warde, P.; Mirimanoff, R.O.; et al. Biopsy diagnosis of intraductal carcinoma is prognostic in intermediate and high risk prostate cancer patients treated by radiotherapy. Eur. J. Cancer 2012, 48, 1318–1325.
- Haroon, U.M.; O’Grady-Coyne, S.; Davis, N.F.; Gullmann, C.; Forde, J.C.; Smyth, G.P.; Power, R.E.; Cheema, I.A.; McLornan, L. Intraductal carcinoma of the prostate in an Irish prostate cancer patient cohort—An aggressive pathology and a strong familial link. Prostate Int. 2020, 8, 107–111.
- Macrini, S.; Francesconi, S.; Caprera, C.; Lancia, D.; Corsi, M.; Gunnellini, M.; Rocchi, A.; Pireddu, A.; Marziani, F.; Mosillo, C.; et al. Looking for a Simplified Diagnostic Model to Identify Potentially Lethal Cases of Prostate Cancer at Initial Diagnosis: An ImGO Pilot Study. Cancers 2022, 14, 1542.
- Risbridger, G.P.; Taylor, R.A.; Clouston, D.; Sliwinski, A.; Thorne, H.; Hunter, S.; Li, J.; Mitchell, G.; Murphy, D.; Frydenberg, M.; et al. Patient-derived Xenografts Reveal that Intraductal Carcinoma of the Prostate Is a Prominent Pathology in BRCA2 Mutation Carriers with Prostate Cancer and Correlates with Poor Prognosis. Eur. Urol. 2015, 67, 496–503.
- Lozano, R.; Salles, D.C.; Sandhu, S.; Aragón, I.M.; Thorne, H.; López-Campos, F.; Rubio-Briones, J.; Gutierrez-Pecharroman, A.M.; Maldonado, L.; di Domenico, T.; et al. Association between BRCA2 alterations and intraductal and cribriform histologies in prostate cancer. Eur. J. Cancer 2021, 147, 74–83.
- Khani, F.; Wobker, S.E.; Hicks, J.L.; Robinson, B.D.; Barbieri, C.E.; De Marzo, A.M.; Epstein, J.I.; Pritchard, C.C.; Lotan, T.L. Intraductal carcinoma of the prostate in the absence of high-grade invasive carcinoma represents a molecularly distinct type of in situ carcinoma enriched with oncogenic driver mutations. J. Pathol. 2019, 249, 79–89.
- Zhu, S.; Zhao, J.; Nie, L.; Yin, W.; Zhang, Y.; Zhao, F.; Ni, Y.; Zhang, X.; Wang, Z.; Dai, J.; et al. Homologous recombination deficiency (HRD) score in aggressive prostatic adenocarcinoma with or without intraductal carcinoma of the prostate (IDC-P). BMC Med. 2022, 20, 237.
- Montironi, R.; Zhou, M.; Magi-Galluzzi, C.; Epstein, J.I. Features and Prognostic Significance of Intraductal Carcinoma of the Prostate. Eur. Urol. Oncol. 2018, 1, 21–28.
- Kweldam, C.F.; van der Kwast, T.; van Leenders, G.J. On cribriform prostate cancer. Transl. Androl. Urol. 2018, 7, 145–154.
- Testa, U.; Castelli, G.; Pelosi, E. Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications. Medicines 2019, 6, 82.
- Rubin, M.A.; de La Taille, A.; Bagiella, E.; Olsson, C.A.; O’Toole, K.M. Cribriform carcinoma of the prostate and cribriform prostatic intraepithelial neoplasia: Incidence and clinical implications. Am. J. Surg. Pathol. 1998, 22, 840–848.
- Keefe, D.T.; Schieda, N.; El Hallani, S.; Breau, R.H.; Morash, C.; Robertson, S.J.; Mai, K.T.; Belanger, E.C.; Flood, T.A. Cribriform morphology predicts upstaging after radical prostatectomy in patients with Gleason score 3 + 4 = 7 prostate cancer at transrectal ultrasound (TRUS)-guided needle biopsy. Virchows Arch. 2015, 467, 437–442.
- Tzelepi, V.; Grypari, I.M.; Logotheti, S.; Stavros Kontogiannis, P.K.; Melachrinou, M.; Zolota, V. Contemporary Grading of Prostate Cancer: The Impact of Grading Criteria and the Significance of the Amount of Intraductal Carcinoma. Cancers 2021, 13, 5454.
- Kweldam, C.F.; Wildhagen, M.F.; Steyerberg, E.W.; Bangma, C.H.; van der Kwast, T.H.; van Leenders, G.J.L.H. Cribriform growth is highly predictive for postoperative metastasis and disease-specific death in Gleason score 7 prostate cancer. Mod. Pathol. 2015, 28, 457–464.
- O’Brien, C.; True, L.D.; Higano, C.S.; Rademacher, B.L.S.; Garzotto, M.; Beer, T.M. Histologic changes associated with neoadjuvant chemotherapy are predictive of nodal metastases in patients with high-risk prostate cancer. Am. J. Clin. Pathol. 2010, 133, 654–661.
- Iczkowski, K.A.; Torkko, K.C.; Kotnis, G.R.; Wilson, R.S.; Huang, W.; Wheeler, T.M.; Abeyta, A.M.; La Rosa, F.G.; Cook, S.; Werahera, P.N.; et al. Digital quantification of five high-grade prostate cancer patterns, including the cribriform pattern, and their association with adverse outcome. Am. J. Clin. Pathol. 2011, 136, 98–107.
- Hollemans, E.; Verhoef, E.I.; Bangma, C.H.; Rietbergen, J.; Osanto, S.; Pelger, R.C.M.; van Wezel, T.; van der Poel, H.; Bekers, E.; Helleman, J.; et al. Cribriform architecture in radical prostatectomies predicts oncological outcome in Gleason score 8 prostate cancer patients. Mod. Pathol. 2021, 34, 184–193.
- Siadat, F.; Sykes, J.; Zlotta, A.R.; Aldaoud, N.; Egawa, S.; Pushkar, D.; Kuk, C.; Bristow, R.G.; Montironi, R.; Van Der Kwast, T. Not all gleason pattern 4 prostate cancers are created equal: A study of latent prostatic carcinomas in a cystoprostatectomy and autopsy series. Prostate 2015, 75, 1277–1284.
- Destouni, M.; Lazaris, A.C.; Tzelepi, V. Cribriform Patterned Lesions in the Prostate Gland with Emphasis on Differential Diagnosis and Clinical Significance. Cancers 2022, 14, 3041.
- Mota, J.M.; Barnett, E.; Nauseef, J.T.; Nguyen, B.; Stopsack, K.H.; Wibmer, A.; Flynn, J.R.; Heller, G.; Danila, D.C.; Rathkopf, D.; et al. Platinum-Based Chemotherapy in Metastatic Prostate Cancer With DNA Repair Gene Alterations. JCO Precis. Oncol. 2020, 4, 355–366.
- Asif, S.; Teply, B.A. Biomarkers for Treatment Response in Advanced Prostate Cancer. Cancers 2021, 13, 5723.
- Risdon, E.N.; Chau, C.H.; Price, D.K.; Sartor, O.; Figg, W.D. PARP Inhibitors and Prostate Cancer: To Infinity and Beyond BRCA. Oncologist 2021, 26, e115–e129.
- Tsujino, T.; Takai, T.; Hinohara, K.; Gui, F.; Tsutsumi, T.; Bai, X.; Miao, C.; Feng, C.; Gui, B.; Sztupinszki, Z.; et al. CRISPR screens reveal genetic determinants of PARP inhibitor sensitivity and resistance in prostate cancer. Nat. Commun. 2023, 14, 252.
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708.
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102.
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men with Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772.
- Fizazi, K.; Piulats, J.M.; Reaume, M.N.; Ostler, P.; McDermott, R.; Gingerich, J.R.; Pintus, E.; Sridhar, S.S.; Bambury, R.M.; Emmenegger, U.; et al. Rucaparib or Physician’s Choice in Metastatic Prostate Cancer. N. Engl. J. Med. 2023, 388, 719–732.
- Yamada, Y.; Beltran, H. The treatment landscape of metastatic prostate cancer. Cancer Lett. 2021, 519, 20–29.
- Taylor, A.K.; Kosoff, D.; Emamekhoo, H.; Lang, J.M.; Kyriakopoulos, C.E. PARP inhibitors in metastatic prostate cancer. Front. Oncol. 2023, 13, 1159557.
- Pham, M.-T.; Gupta, A.; Gupta, H.; Vaghasia, A.; Skaist, A.; Garrison, M.A.; Coulter, J.B.; Haffner, M.C.; Zheng, S.L.; Xu, J.; et al. Identifying Phased Mutations and Complex Rearrangements in Human Prostate Cancer Cell Lines through Linked-Read Whole-Genome Sequencing. Mol. Cancer Res. 2022, 20, 1013–1020.
- Liu, Q.; Tong, D.; Liu, G.; Yi, Y.; Xu, J.; Yang, X.; Wang, L.; Zhang, J.; Ye, J.; Zhang, Y.; et al. A novel BRCA2 mutation in prostate cancer sensitive to combined radiotherapy and androgen deprivation therapy. Cancer Biol. Ther. 2018, 19, 669–675.
- Antonarakis, E.S.; Velho, P.I.; Fu, W.; Wang, H.; Agarwal, N.; Santos, V.S.; Maughan, B.L.; Pili, R.; Adra, N.; Sternberg, C.N.; et al. CDK12-Altered Prostate Cancer: Clinical Features and Therapeutic Outcomes to Standard Systemic Therapies, Poly (ADP-Ribose) Polymerase Inhibitors, and PD-1 Inhibitors. JCO Precis. Oncol. 2020, 4, 370–381.
- Schweizer, M.T.; Ha, G.; Gulati, R.; Brown, L.C.; McKay, R.R.; Dorff, T.; Hoge, A.C.H.; Reichel, J.; Vats, P.; Kilari, D.; et al. CDK12 -Mutated Prostate Cancer: Clinical Outcomes with Standard Therapies and Immune Checkpoint Blockade. JCO Precis. Oncol. 2020, 4, 382–392.
- Marshall, C.H.; Sokolova, A.O.; McNatty, A.L.; Cheng, H.H.; Eisenberger, M.A.; Bryce, A.H.; Schweizer, M.T.; Antonarakis, E.S. Differential Response to Olaparib Treatment Among Men with Metastatic Castration-resistant Prostate Cancer Harboring BRCA1 or BRCA2 Versus ATM Mutations. Eur. Urol. 2019, 76, 452–458.
- Gillette, C.M.; Yette, G.A.; Cramer, S.D.; Graham, L.S. Management of Advanced Prostate Cancer in the Precision Oncology Era. Cancers 2023, 15, 2552.
- Goodall, J.; Mateo, J.; Yuan, W.; Mossop, H.; Porta, N.; Miranda, S.; Perez-Lopez, R.; Dolling, D.; Robinson, D.R.; Sandhu, S.; et al. Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov. 2017, 7, 1006–1017.
- Deluce, J.E.; Cardenas, L.; Lalani, A.-K.; Vareki, S.M.; Fernandes, R. Emerging Biomarker-Guided Therapies in Prostate Cancer. Curr. Oncol. 2022, 29, 5054–5076.
- Mateo, J.; McKay, R.; Abida, W.; Aggarwal, R.; Alumkal, J.; Alva, A.; Feng, F.; Gao, X.; Graff, J.; Hussain, M.; et al. Accelerating precision medicine in metastatic prostate cancer. Nat. Cancer 2020, 1, 1041–1053.
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020, 34, 360–394.
- Antonarakis, E.S.; Gomella, L.G.; Petrylak, D.P. When and How to Use PARP Inhibitors in Prostate Cancer: A Systematic Review of the Literature with an Update on On-Going Trials. Eur. Urol. Oncol. 2020, 3, 594–611.
- Nientiedt, C.; Tolstov, Y.; Volckmar, A.-L.; Endris, V.; Bonekamp, D.; Haberkorn, U.; Jäger, D.; Sültmann, H.; Stenzinger, A.; Hohenfellner, M.; et al. PARP inhibition in BRCA2-mutated prostate cancer. Ann. Oncol. 2017, 28, 189–191.
More