Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by François-Xavier Campbell-Valois and Version 2 by Fanny Huang.
Shigella spp. cause hundreds of millions of intestinal infections each year. They target the mucosa of the human colon and are an important model of intracellular bacterial pathogenesis. Shigella is a pathovar of Escherichia coli that is characterized by the presence of a large invasion plasmid, pINV, which encodes the characteristic type III secretion system and icsA used for cytosol invasion and cell-to-cell spread, respectively.
- Shigella
- Escherichia coli
- pathogenesis
- plasmid
1. Introduction
Many gram-negative bacteria, such as Bordetella, Burkholderia, Citrobacter, Chlamydia, Escherichia, Pseudomonas, Rhizobium, Salmonella, Shigella, Xanthomonas, and Yersinia, interact with host cells using the syringe-shaped type III secretion system (T3SS) also known as the injectisome. This large proteinaceous complex injects substrate proteins into host cells to hijack them for the benefit of the bacteria that harbor it. Since 2015, spectacular progress has been made in describing its structure and function [1][2][3][4][5][6][7][8][9][1,2,3,4,5,6,7,8,9]. The T3SS is composed of the sorting platform that selects the protein substrates in the bacterial cytosol, the transmembrane needle complex that serves as a channel for the secretion of the substrates, the tip complex that triggers secretion upon sensing the physical contact with host cells, and the translocon that establishes the continuity between the needle complex and the host cytosol during the delivery of the substrates [7]. The Shigella flexneri T3SS has been one of the most extensively studied, providing us with an in-depth understanding of its role and function in pathogenesis (reviewed in [10][11][12][10,11,12]). In Shigella, the fate of the T3SS is tied to the large invasion plasmid that encodes it. Acquisition of this plasmid drove the evolution of Shigella spp. from Escherichia coli. This makes Shigella a powerful model to study the evolution of virulence and the interplay of the T3SS with its genome.
2. The Evolution of the Shigella Pathovar
Shigella spp. are gram-negative enterobacteria divided in four subgroups named S. boydii, S. dysenteriae, S. flexneri, and S. sonnei. Their unique pathogenesis, their lack of flagellar motility and inability to ferment lactose constitutes the hallmark of the Shigella genus established in the 1950s [13]. However, modern phylogeny approaches revealed that Shigella spp. are merely an Escherichia coli pathovar [14]. Shigella spp. infect the large intestine of humans and cause diarrheal symptoms ranging from watery to mucopurulent and bloody stools accompanied by inflammation (also known as dysentery). Shigella spp. cause 80–190 million infections [15][16][15,16], and approximately 200,000 deaths annually, with more than 50% of cases occurring in individuals younger than 5 years or older than 70 years [17]. S. flexneri is responsible for most cases and, similar to S. dysenteriae, is more common in low-income countries. By contrast, S. sonnei is responsible for most cases in high-income countries. [18]. Colony-forming units in the low hundreds can cause the disease in healthy humans [19], making Shigella more infectious than most other enterobacteria. Shigella has no known animal reservoirs and is therefore considered to be human specific. It is transmitted from person to person or by ingestion of contaminated water or food. As with other enterobacteria [20][21][22][23][24][20,21,22,23,24], increasing antibiotic resistance in Shigella spp. is a public health concern [20][25][26][27][20,25,26,27].
From an evolutionary perspective, Shigella emerged 35,000–270,000 years ago [28][29][28,29], contemporaneously with Homo sapiens. The evolutionary trajectories of S. dysenteriae, S. flexneri, and S. sonnei. are unique [30][31][32][30,31,32]. In the case of S. dysenteriae and S. sonnei, modern historical events and societal changes have greatly accelerated their expansion and spread. Shigella spp. have adapted to their human host by losing or inactivating chromosomal genes detrimental to pathogenesis [33][34][33,34]. Thus, the number of chromosomal genes in Shigella is reduced compared to their commensalistic E. coli counterparts, a phenomenon shared with several bacterial pathogens. In a remarkable example of convergent evolution, some Shigella strains lost the same chromosomal genes through disruption by discrete insertion sequences [28][35][28,35]. Thus, an intriguing question about Shigella is whether it emerged once [36], or on multiple occasions [28] within the E. coli lineage. Since these seminal studies, the number of sequenced E. coli genomes has increased dramatically, allowing further study of their phylogeny [37][38][39][37,38,39]. For example, analysis of 10,667 chromosome sequences, including 1283 from Shigella spp., using the fast distance estimation method MASH revealed 14 E. coli phylogroups [37]. Two of these, named Shig1 and Shig2, consisted exclusively of Shigella strains, with an overrepresentation of S. flexneri and S. sonnei, respectively, and contained most of the Shigella genomes in the dataset (Table 1). This analysis confirmed on an unprecedented scale that Shigella shared several of the hallmarks of bacterial pathogens. Indeed, Shig1 and Shig2 strains had a lower guanine-cytosine content, smaller genomes, and higher rates of gene loss and duplication on average than other E. coli phylogroups. The analyses also revealed that S. sonnei in Shig2 possesses a unique set of core genes that are not conserved in other phylogroups, highlighting its clonality and unique evolutionary origin. Nevertheless, Shigella strains were also found in seven other phylogroups (Table 1), with B1 being the most dominant. Interestingly, analyses measuring the conservation and loss of protein families confirmed the remarkable convergent evolution of Shig1 and Shigella from the B1 phylogroup.
Table 1. Distribution of Shigella strains in E. coli phylogroups 1.
| Phylogroups | Total Sequences |
Shigella | Sequences |
Percentage | Shigella | in Group (%) | Percentage of Total | Shigella | (%) |
|---|---|---|---|---|---|---|---|---|---|
| All | 10,667 | 1283 | 12.0 | 100 | |||||
| Shig1 | 177 | 177 | 100 | 70.1 | |||||
| Shig2 | 899 | 899 | 100 | 13.8 | |||||
| B1 | 2960 | 140 | 4.73 | 10.9 | |||||
| A | 2232 | 44 | 1.97 | 3.43 | |||||
| E1 | 279 | 9 | 3.23 | 0.70 | |||||
| D3 | 177 | 7 | 3.95 | 0.55 | |||||
| D2 | 177 | 4 | 2.26 | 0.31 | |||||
| F | 199 | 2 | 1.01 | 0.16 | |||||
| B2-2 | 1367 | 1 | 0.07 | 0.08 | |||||
| G | 96 | 0 | 0 | 0 | |||||
| D1 | D1 | 0 | 0 | 0 | |||||
| C | 540 | 0 | 0 | 0 | |||||
| B2-1 | 541 | 0 | 0 | 0 |
1 The data in this table are from the Supplementary Information from Abram et al. [37].
The evolution and pathogenesis of Shigella spp. and of the less virulent enteroinvasive E. coli (EIEC) is associated with the acquisition of a large invasion plasmid of 220 kbp, called pINV, which has a high degree of sequence conservation across strains [30][40][41][42][43][30,40,41,42,43]. The genome remodeling in EIECs is less pronounced than in Shigella. Therefore, EIECs can be described as an intermediate between commensal E. coli and Shigella, which may help us to understand the evolution of the Shigella-EIEC pathotype and the coevolution of the chromosome with pINV [42]. Since pINV played a key role in the emergence of Shigella and EIEC clades, each of them should have originated from an independent pINV acquisition event (Figure 1a). Although a consensus has emerged around this model [28][29][37][44][28,29,37,44], the fact that pINV is not conjugative, and therefore not susceptible to horizontal transfers, is a hurdle that, to researchers' knowledge, has not yet been addressed. The alternative model proposes that pINV was acquired once by a common ancestor of all Shigella and a subset of E. coli [36] (Figure 1b). Shigella would have conserved pINV by adapting their genome to the pathogenic lifestyle, whereas non-EIEC E. coli would have lost it by failing to make these adaptations. This model accounts for the non-transmissibility of pINV, but it has its own set of challenges. Not least of these is that it requires the relatively unstable pINV to have been maintained over the evolutionary time required for the emergence of Shigella spp. Despite these limitations, researchers believe that the single acquisition model, or a hybrid model in which related Shigella clades may be the result of a single pINV acquisition event, should be considered in light of the large number of new sequences obtained since it was first described [36].
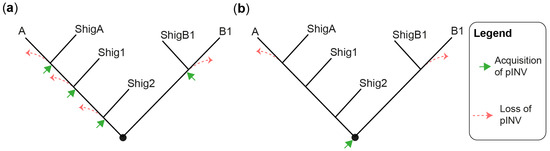
Figure 1. Models for the emergence of Shigella from E. coli. (a) A tree illustrating the multiple pINV acquisition model. (b) A tree illustrating the single pINV acquisition model. These phylogenetic trees are qualitative and intended to be used for the sole purpose of illustrating the main difference between the two models in a straightforward manner. ShigA: Shigella from phylogroup A; ShigB1: Shigella from phylogroup B1; Shig1 and Shig2 as defined in the text and Table 1.