Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Fanny Huang and Version 1 by Peng WANG.
With an increase in energy consumption globally, Fischer-Tropsch (FT) synthesis is a good alternative for producing fuels and chemicals from coal, natural gas or biomass. Among them, coal to liquids has been put into production in countries that have large coal reserves. In this process, Fe-based catalysts are commonly used due to their earth abundance, comparatively wide operation range and ready availability to handle low H2/CO ratio from coal.
- Fischer-Tropsch synthesis
- Fe-carbide
- catalysts
- Fe-based catalysts
1. Fischer-Tropsch Synthesis
Fischer-Tropsch (FT) process has been widely investigated for almost 100 years. In the 1920s, German scientists Franz Fischer and Hans Tropsch first developed this process, in which a mixture of H2 and CO (synthesis gas) can be converted to valuable long-chain hydrocarbons like gasoline, diesel fuel and chemicals (olefins, alcohols or acids) [1,2][1][2]. The FT process was first commercialized in Germany prior to the Second World War. It could offer synthetic fuel for the German war machine due to the abundant domestic coal supplies used for producing synthesis gas [3]. After the Second World War, the development of this process stalled because low crude oil prices led to a strong growth and dominance of the petroleum oil industry [4]. The interest in the FT synthesis was revived in South Africa during the Apartheid regime in the 1970s. During this period, the supply of oil in South Africa was cut off due to international sanctions, but through the FT synthesis, South Africans were still able to produce the required fuels and chemicals from coal. At the same time, the energy crises in 1973 and 1978 have also stimulated the global interest and exploration of alternative fuel production by expanding the commercialization of FT processes [5]. Besides coal, natural gas and biomass are also considered as important alternatives. The process to convert these carbon sources to valuable chemicals is often referred to as “X to liquid” (XTL), in which X stands for the feedstock from which synthesis gas is derived, e.g., coal (CTL), natural gas (GTL) or biomass (BTL) [6]. Although most of these embodiments still rely on fossil resources, CTL and GTL enable the production of clean transportation fuels that are free of heavy metals, aromatics or contaminants such as nitrogen and sulfur. Currently, large XTL processes are operated in Malaysia by Shell, in Qatar by Shell and SASOL, and in South Africa, Uzbekistan and Nigeria by SASOL. Among them, CTL is of large interest in areas with abundant coal resources, for example China and South Africa. Currently, a large number of CTL demonstration plants and industrialization projects are commissioned in China using Fe-based catalysts [7].
The CTL process generally consists of four chemical conversion steps (Figure 1). In the first step, coal gasification is performed in gasifiers to produce synthesis gas, a mixture of CO and H2. Due to the low H/C ratio of coal, the derived synthesis gas has a typical H2/CO ratio below 1 [8]. In the second step, the water-gas shift (WGS) reaction (Equation (1)) is used to increase the H2/CO ratio tailored for the desired product distribution in the subsequent FT synthesis step. Usually, a constant amount of CO2 is removed in the overall CTL process, either in a WGS step prior to the FT synthesis or in the FT synthesis reactor itself when a catalyst is used that exhibits sufficient WGS activity. CO2 produced in the FT reactor not only decreases CO conversion, but also leads to a higher energy consumption for separating the gas effluent from the reactor. When the main WGS conversion is conducted in a separate WGS reactor, CO2 capture becomes more viable [9]. After cooling and purification, the synthesis gas is introduced into the FT synthesis reactor and converted into long-chain hydrocarbons (Equation (2)). Traditionally, these processes are realized in either fixed and fluidized bed reactors or slurry bubble reactors. Fixed beds are suitable for wax production, as for instance is conducted in Shell FT plants in Malaysia and Qatar [10]. The separation of products from catalysts are more cost-effective in fixed bed reactors. On the other hand, the pressure drop in such reactors leads to higher operational costs than other reactors. Moreover, as it is costly to replace the catalyst inventory, catalysts should exhibit a long lifespan. As the FT reaction is highly exothermic, it is important to rapidly remove the heat of reaction in order to avoid overheating the catalyst [11]. Compared to fixed bed reactors, fluidized bed reactors can realize a very homogeneous temperature distribution because of the rapid and turbulent gas/liquid movement. Another important advantage of fluidized bed reactors is that a deactivated catalyst can be removed from the bottom of the reactors by gas flushing, and new catalysts can be added to replenish the spent ones for longer production runs. A drawback of such reactors is the difficulty in separating the catalyst from the products. Fluidized bed reactors are considered to be a promising technology for the production of lower-molecular-weight products on Fe-based catalysts at high temperature [12]. Similar to fluidized bed reactors, slurry bubble reactors can meet the requirement of an online removal/addition of a catalyst and can be operated under isothermal conditions. Catalysts that feature high mechanical strength and attrition resistance are required for slurry bubble reactors. As the catalysts are suspended in the wax, separating the catalyst from wax is a major challenge [13].
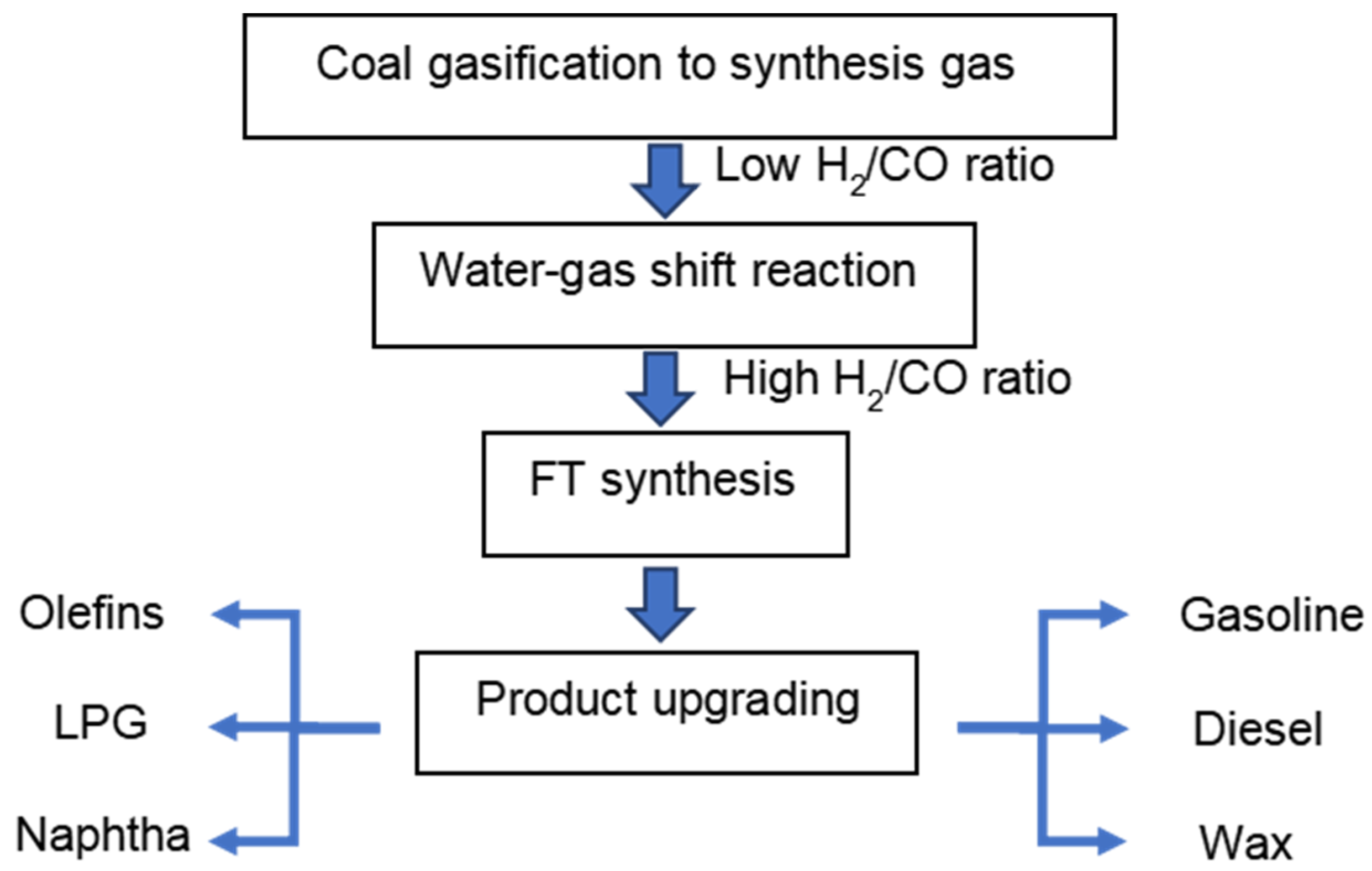
Figure 1. Schematic representation of the CTL process.
The final step of the overall XTL process is product upgrading. The mixture of products formed during the FT process (e.g., long-chain hydrocarbons or oxygenates) need to be processed in order to obtain high-value transportation fuels and base chemicals using processes such as hydrotreating, hydrocracking and hydro-isomerization. The in-reactor upgrading of the products of FT reactions by adding zeolites to Fe-based catalysts has also been investigated [14,15][14][15]. Among a variety of hydrocarbons, linear α-olefins (LAOs) production and separation are gaining widespread attractions. LAOs are highly valuable intermediates for the chemical industry [16]. Lower olefins (C2–C4 LAOs) are mainly used as building blocks, commonly produced by the steam cracking of ethane or naphtha and the dehydrogenation of propane [17]. The process to produce lower olefins via FT reaction is referred to as “Fischer-Tropsch to olefins” (FTO). A substantial amount of work has been conducted to develop highly efficient catalysts to directly convert synthesis gas to lower olefins [18,19][18][19]. However, traditional FTO is limited by ASF distribution and suffers from high CH4 selectivity. To address these issues, recently, the development of oxide–zeolite bifunctional catalysts to selectively convert synthesis gas to lower olefins has seen significant progress. The separation of CO activation and C–C coupling onto two different types of active sites can tune C2–C4 LAOs selectivity as high as 80% at a fair CO conversion [20,21][20][21]. LAOs with more than four carbon atoms, especially in the C5–C10 range, are even more valuable than lower olefins because of their use as co-monomers in polymerization and as feedstocks for lubricants and detergents [22]. Currently, there remains no commercial process for directly converting synthesis gas to higher LAOs which do not meet consumer demand, thus hampering industrial development globally.
CO + H2O → CO2 + H2
nCO + (2n + 1)H2 → CnH2n+2 + nH2O
2. Catalysts
A general aspect of FT chemistry is the dissociation of CO in atomic C and O on metal surfaces [23], although there are also pathways that involve the hydrogenation of CO prior to C–O bond cleavage. Group VIII metals with unoccupied d-orbitals are capable of CO dissociation. Based on Brønsted-Evans-Polanyi (BEP) relations, transition metals with a lower d-band filling that will bind the dissociated C and O atoms strongly will lead to low activation barriers for CO dissociation [24]. The termination of the chain-growth reactions are also important as they determine the length of the hydrocarbons obtained [23].
From left to right in the periodic table, the d band of transition metals is filled [25]. Catalysts with a lower C and O binding energy to the right will hardly produce long-chain hydrocarbons, because the CHx growth monomers are easily hydrogenated by H2, resulting in high CH4 selectivity. Therefore, CO hydrogenation on Ni mainly produces CH4. For Cu- and Rh-based catalysts, the main products for CO hydrogenation are alcohols due to their low CO dissociation ability. Metals such as Ru, Co and Fe bind C and O stronger than Ni, Cu and Rh, resulting in a higher probability for CHx intermediate to couple to long-chain hydrocarbons. Hence, Ru-, Co- and Fe-based catalysts are the most suitable for the FT reaction [26]. The product distribution of CO hydrogenation on transition metals is shown in Figure 2.
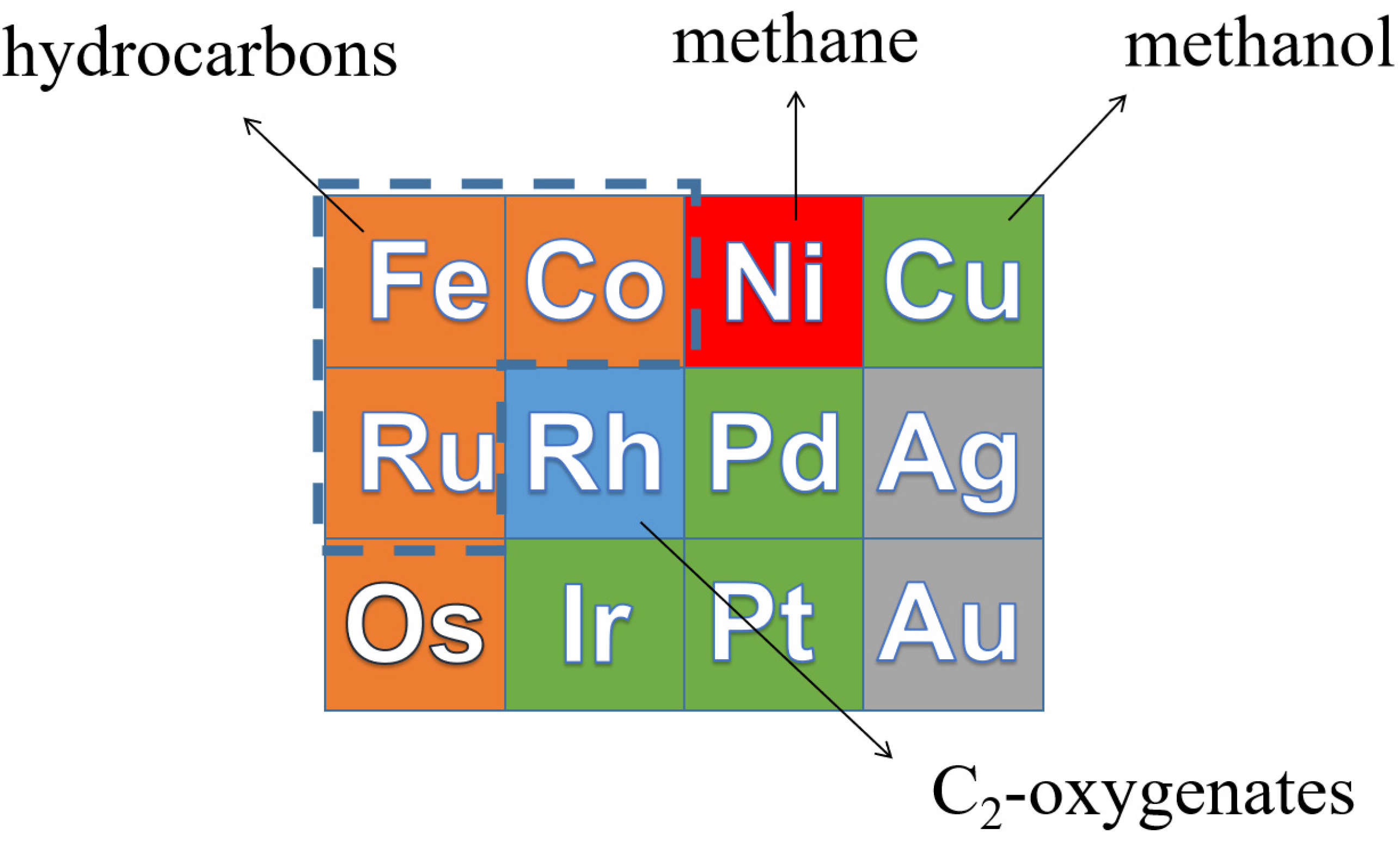
Figure 2. The product distribution of CO hydrogenation on transition metals.
Ru-based catalysts display outstanding performance for CO hydrogenation in terms of activity, selectivity and stability. Despite this, Ru cannot serve as a base for catalysts at the industrial scale because of the high price of this cheapest of noble metals [27]. So far, only Co and Fe have been used as the active phase for industrial FT catalysts.
Co-based catalysts outperform Fe-based catalysts at low temperature (200–240 °C), often referred to as low-temperature Fischer-Tropsch (LTFT). Moreover, the WGS activity of Co is much lower than that of Fe, limiting undesired CO2 formation. On the other hand, the higher CH4 selectivity on Co-based catalysts restricts its application at high temperature (250–350 °C), which is referred to as high-temperature Fischer-Tropsch (HTFT). In HTFT, Fe-based catalysts are preferred to reduce the amount of CH4 formed [17]. Another advantage of Fe catalysts is that they can handle the low H2/CO ratio of synthesis gas derived from coal and biomass, owing to the substantial WGS activity [28]. On the other hand, Co-based catalysts are typically used in combination with natural gas as a source of the synthesis gas feedstock. Overall, the operation conditions for Fe-based catalysts are more flexible and they can also be used in LTFT. Paraffins, such as wax, is the main product for Co-based catalyst in LTFT, whereas Fe is mainly used for producing olefins and oxygenates in HTFT. Metallic Co is regarded as the active phase for the FT reaction, while Fe has a high tendency to form Fe-carbide because of a strong Fe–C bond. The most significant differences between Co- and Fe-based catalysts are shown in Table 1.
| Property | Co | Fe |
|---|---|---|
| Cost | expensive | cheap |
| Reaction temperature | 200–240 °C | 250–350 °C |
| FT activity | high | relativley low |
| WGS activity | neglible | active |
| Carbon source | Natural gas | Coal and biomass |
| H2/CO ratio | ~2 | 0.5–2.5 |
| Active phase | Metallic Co | Fe-carbides |
| Methane selectivity | high | low |
| Products | Wax (paraffins) | C1–C15, olefins, oxygenates |
| Sulfur tolerance | Very sensitive | sensitive |
In addition to mono-Co or Fe-based catalysts, the construction of bimetallic catalysts incorporating both Co and Fe has attracted a wide range of attention. Yang et al. revealed that adding Co in χ-Fe5C2 enhances the FT performance because Co is more capable of dissociating CO [31]. The synergistic effect of Co–Fe alloy not only leads to a higher FT activity, but is also conducive to grow long-chain hydrocarbons [32]. It was also reported that bimetallic catalysts were more stable against deactivation compared to pure Co-based catalysts, even though they still suffered from deactivation at high CO conversion due to high H2O partial pressure [33].
3. Fe-Based Catalysts
In the Earth’s crust, Fe is the fourth most abundant element, mainly existing in the form of Fe-oxide. This abundance means that Fe is very cheap and an excellent choice for the FT catalysis. For the industrial FT synthesis, precipitated or fused Fe in unsupported form are mainly used as catalyst precursors [34]. Practically, precipitated Fe is employed in fixed bed or slurry bed reactor in LTFT, predominately producing long-chain hydrocarbons, e.g., wax [35]. On the other hand, fused Fe is mainly consumed in fluidized beds in HTFT for olefins production [12]. The poor mechanical strength of unsupported catalysts may lead to the plugging of the catalyst bed in fixed bed operation or to the fouling of downstream equipment in fluidized bed operation. Supported Fe catalysts display an enhanced dispersion of the active phase and may withstand the mechanical degradation that threatens unsupported catalysts.
Upon activation, the Fe-oxide precursor is usually converted into a mixture of metallic Fe, Fe-carbides and Fe-oxides and the composition of catalyst depends on many parameters such as the catalyst precursor, catalyst pretreatment and the FT reaction conditions [36]. Despite the complexity of the composition in the working condition, a correlation between Fe-carbide content and FT activity has been widely observed and Fe-carbide formation is believed to be the necessary step to obtain good FT activity [37]. Fe-oxide, on the other hand, is considered to be active for the WGS reaction, which leads to the production of excessive CO2 [38]. In itself, CO2 production represents a loss of valuable carbon products. As some WGS operation is needed in CTL and BTL processes, it can be worthwhile to remove CO2 to reduce the greenhouse gas emissions of the process. A distinct feature of Fe-carbide is that it is air sensitive [39] and readily oxidized in air at room temperature, leading to the formation of Fe-oxide [40]. Thus, activation or carbide formation is preferably performed in situ before starting the FT reaction [41]. For research purposes, the passivation of the catalyst in diluted O2 is usually employed [42].
During FT reaction, ε(’)-carbide, χ-Fe5C2 and Θ-Fe3C are commonly observed in Fe-based FT catalysts [43,44][43][44]. They are classified as interstitial carbides, because C atoms occupy the interstices of the Fe lattice. According to the way in which C atoms occupy the hexagonally close-packed (hcp) lattice, their structure can be divided into two categories. In ε(’)-carbide, C atoms occupy Fe octahedral interstices ascribed to octahedral carbides, while the C atoms in Θ-Fe3C and χ-Fe5C2 are situated in trigonal prismatic interstices. The main differences between these three Fe-carbides are listed in Table 2.
Table 2. Comparison of ε(′)-carbide, χ-Fe5C2 and Θ-Fe3C.
| ε(′)-Carbide | χ-Fe5C2 | Θ-Fe3C | |
|---|---|---|---|
| Space group | P63/mmc | C2/c | pnma |
| Crystal structure | hexagonal | monoclinic | orthorhombic |
| C/Fe | 0.45~0.5 | 0.4 | 0.33 |
| Required carbon chemical potential | High (low T, high H2/CO) |
Low (high T, low H2/CO) |
Low (high T, low H2/CO) |
Among these three carbides, ε(′)-carbide is the generic term for ε-Fe2C and ε’-Fe2.2C. ε-Fe2C and ε’-Fe2.2C share the same space group and lattice parameter, but differ in the chemical environment of Fe atoms [45]. The site occupancy of C in ε-Fe2C and ε′-Fe2.2C is 0.5 and 0.45, respectively. ε(′)-carbide formation is favored at a high carbon chemical potential, which represents the condition of low temperature and high CO partial pressure [36]. However, kinetic factors (lattice deformation, carbon diffusion) can prevent its formation at low temperature. Hence, they are commonly observed in catalysts with relatively small particles and in the presence of a support material or chemical promoters [46,47][46][47]. If temperature exceeds 250 °C, ε(′)-carbide will transform to χ-Fe5C2 [48,49][48][49]. χ-Fe5C2 is the most observed carbide phase in the context of FT catalysis. Several works show that χ-Fe5C2 is the main active phase constituent at moderate FT conditions, owing to its relative thermodynamic stability at low carbon chemical potential. When the temperature is further increased, χ-Fe5C2 will transform to Θ-Fe3C [50]. The less active Θ-Fe3C can also evolve into a more active χ-Fe5C2 [51]. The common feature of carbon-poor Θ-Fe3C is that it can contribute to the buildup of carbonaceous deposits because of its near-metallic nature [36,52][36][52]. Excessive carbonaceous deposits like graphite will cause the deactivation of the catalyst [53,54][53][54].
Despite extensive research, it is still uncertain which phase is the most active in the FT reaction. The intrinsic activity comparison between ε(′)-carbide and χ-Fe5C2 is widely investigated. Usually, a mixture of several Fe-carbides is obtained during reaction, making it impossible to correlate activity with a specific phase [36,55][36][55]. Chang et al. found that the intrinsic activity of χ-Fe5C2 is higher than ε(′)-carbide through changing the pretreatment condition [56]. However, Chun et al. found that ε(′)-carbide performs better than χ-Fe5C2 by introducing CO2 in the feed, which increased the amount of ε(′)-carbide [57]. Lu et al. also found that the content of ε(′)-carbide is more active than χ-Fe5C2 [58]. Another exploration by Wezendonk et al. pointed out that the weight-normalized activities (FTY) of χ-Fe5C2 and ε(′)-carbide are virtually identical [59]. There are also some controversial conclusions in terms of the product distribution on different Fe-carbide phases within an FT reaction [36,55,59,60][36][55][59][60]. The observed differences are probably caused by various factors, such as particle size, exposed facet (phase morphology), surface carbon deposition, support effect and the interference of other phases. To overcome these difficulties, surface-sensitive techniques were applied to in situ measure the chemical and structural composition of well-defined catalysts (single-crystal or flat-model catalysts) under operating conditions [61,62][61][62]. These systems were mainly focused on studying time-, pressure-, temperature-dependent Fe carburization kinetics. To further establish a structure-activity relationship on Fe-carbide in the FT reaction and study the underlying mechanism, synthesizing specific pure Fe-carbide and realizing that the catalyst suffers from no phase change or deactivation during FT reaction is of paramount importance.
Some endeavors have been made to synthesize oxide-free Fe-carbides. By using a rapid quenched skeletal iron precursor, Xu et al. prepared a catalyst that mainly consists of ε(′)-carbide and it showed excellent FT activity at relatively low temperature [45]. Peng et al. took advantage of Raney Fe, which is porous and support-free and can be used as a model catalyst to successfully synthesize phase-pure ε(′)-carbide by tuning carburization conditions. The obtained ε(′)-carbide showed low CO2 selectivity in LTFT reaction [9]. The formed ε(′)-carbide is also stable at higher temperature and the phase composition does not change after operating at 250 °C. By stabilizing ε(’)-carbide into graphene layers, it can also catalyze HTFT reaction without transforming to χ-Fe5C2 [63]. Single-phase χ-Fe5C2 was synthesized by Yang et al. using a facile wet-chemical route with the help of a Br agent [64]. In their following work, a range of phase-pure metallic Fe and Fe-carbide nanoparticles were utilized in illustrating the FT mechanism [60].
References
- Fischer, F.; Tropsch, H. The Synthesis of Petroleum at Atmospheric Pressures from Gasification Products of Coal. Brennst. Chem. 1926, 7, 97–104.
- Fischer, F.; Tropsch, H. Development of Noval Catalysts for Fischer-Tropsch Synthesis. Brennst. Chem. 1923, 4, 276–285.
- Office of Fossil Energy and Carbon Management. Early Days of Coal Research. 2000. Available online: https://www.energy.gov/fecm/early-days-coal-research (accessed on 5 April 2022).
- Zhang, Q.; Kang, J.; Wang, Y. Development of Novel Catalysts for Fischer-Tropsch Synthesis: Tuning the Product Selectivity. ChemCatChem 2010, 2, 1030–1058.
- de Smit, E.; Weckhuysen, B.M. The Renaissance of Iron-based Fischer-Tropsch Synthesis: On the Multifaceted Catalyst Deactivation Behaviour. Chem. Soc. Rev. 2008, 37, 2758–2781.
- King, D.L.; de Klerk, A. Overview of Feed-to-Liquid (XTL) Conversion. ACS Symp. Ser. 2011, 1084, 1–24.
- Hao, X.; Dong, G.; Yang, Y.; Xu, Y.; Li, Y. Coal to Liquid (CTL): Commercialization Prospects in China. Chem. Eng. Technol. 2007, 30, 1157–1165.
- Cao, Y.; Gao, Z.; Jin, J.; Zhou, H.; Cohron, M.; Zhao, H.; Liu, H.; Pan, W. Synthesis Gas Production with an Adjustable H2/CO Ratio through the Coal Gasification Process: Effects of Coal Ranks and Methane Addition. Energy Fuels 2008, 22, 1720–1730.
- Wang, P.; Chen, W.; Chiang, F.K.; Dugulan, A.I.; Song, Y.; Pestman, R.; Zhang, K.; Yao, J.; Feng, B.; Miao, P.; et al. Synthesis of Stable and Low-CO2 Selective Iron carbide Fischer-Tropsch Catalysts. Sci. Adv. 2018, 4, eaau2947.
- Dry, M.E. The Fischer-Tropsch process: 1950–2000. Catal. Today 2002, 71, 227–241.
- Dry, M.E.; Hoogendoorn, J.C. Technology of the Fischer-Tropsch Process. Catal. Rev. Sci. Eng. 2006, 23, 265–278.
- Steynberg, A.P.; Espinoza, R.L.; Jager, B.; Vosloo, A.C. High Temperature Fischer-Tropsch Synthesis in Commercial Practice. Appl. Catal. A Gen. 1999, 186, 41–54.
- Dry, M.E. Practical and Theoretical Aspects of the Ccatalytic Fischer-Tropsch Process. Appl. Catal. A Gen. 1996, 138, 319–344.
- Karre, A.V.; Kababji, A.; Kugler, E.L.; Dadyburjor, D.B. Effect of Addition of Zeolite to Iron-based Activated-carbon-supported Catalyst for Fischer-Tropsch Synthesis in Separate Beds and Mixed Beds. Catal. Today 2012, 198, 280–288.
- Martínez, A.; López, C. The Influence of ZSM-5 Zeolite Composition and Crystal Size on the in situ Conversion of Fischer-Tropsch Products over Hybrid Catalysts. Appl. Catal. A Gen. 2005, 294, 251–259.
- Tobisch, S.; Ziegler, T. Catalytic Oligomerization of Ethylene to Higher Linear α-Olefins Promoted by Cationic Group 4 Cyclopentadienyl-Arene Active Catalysts: Toward the Computational Design of Zirconium- and Hafnium-Based Ethylene Trimerization Catalysts. Organometallics 2005, 24, 256–265.
- Galvis, H.M.T.; Bitter, J.H.; Khare, C.B.; Ruitenbeek, M.; Dugulan, A.I.; de Jong, K.P. Supported Iron Nanoparticles as Catalysts for Sustainable Production of Lower olefins. Science 2012, 335, 835–838.
- Xu, Y.; Li, X.; Gao, J.; Wang, J.; Ma, G.; Wen, X.; Yang, Y.; Li, Y.; Ding, M. A Hydrophobic Catalyst Increases Olefins from Syngas by Suppressing C1 by-products. Science 2021, 371, 610–613.
- Xie, J.; Palalanen, P.P.; van Deelen, T.W.; Wechhuysen, B.M.; Louwerse, M.J.; de Jong, K.P. Promoted Cobalt Metal Catalysts Suitable for the Production of Lower Olefins from Natural Gas. Nat. Commun. 2019, 10, 167.
- Jiao, F.; Li, J.; Pan, X.; Xiao, J.; Li, H.; Ma, H.; Wei, M.; Pan, Y.; Zhou, Z.; Li, M.; et al. Selective Conversion of Syngas to Light Olefins. Science 2016, 351, 1065–1067.
- Cheng, K.; Gu, B.; Liu, X.; Kang, J.; Zhang, Q.; Wang, Y. Direct and Highly Selective Conversion of Synthesis Gas into Lower Olefins: Design of a Bifunctional Catalyst Combining Methanol Synthesis and Carbon-Carbon Coupling. Angew. Chem. Int. Ed. 2016, 55, 4725–4728.
- Belov, G.P. Tetramerization of Ethylene to Octene1 (A Review). Pet. Chem. 2012, 52, 139–154.
- Filot, I.A.W.; van Santen, R.A.; Hensen, E.J.M. Quantum Chemistry of the Fischer-Tropsch Reaction Catalysed by a Stepped Ruthenium Surface. Catal. Sci. Technol. 2014, 4, 3129–3140.
- Zhao, Z.; Liu, S.; Zha, S.; Cheng, D.; Studt, F.; Henkelman, G.; Gong, J. Theory-guided Design of Catalytic Materials Using Scaling Relationships and Reactivity Descriptors. Nat. Rev. Mater. 2019, 4, 792–804.
- van Bokhoven, J.A.; Miller, J.T. d Electron Density and Reactivity of the d Band as a Function of Particle Size in Supported Gold Catalysts. J. Phys. Chem. C 2007, 111, 9245–9249.
- Schulz, H. Short History and Present Trends of Fischer-Tropsch Synthesis. Appl. Catal. A Gen. 1999, 186, 3–12.
- Ma, W.; Dalai, A.K. Effects of Structure and Particle Size of Iron, Cobalt and Ruthenium Catalysts on Fischer-Tropsch Synthesis. Reactions 2021, 2, 62–77.
- van der Laan, G.P.; Beenackers, A.A.C.M. Kinetics and Selectivity of the Fischer-Tropsch Synthesis: A Literature Review. Catal. Rev. 1999, 41, 255–318.
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Advances in the Development of Novel Cobalt Fischer-Tropsch Catalysts for Synthesis of Long-Chain Hydrocarbons and Clean Fuels. Chem. Rev. 2007, 107, 1692–1744.
- Schulz, H. Comparing Fischer-Tropsch Synthesis on Iron-and Cobalt Catalysts: The Dynamics of Structure and Function, Fischer-Tropsch Sythesis. Catal. Catal. 2007, 163, 177–199.
- Yang, C.; Zhao, B.; Gao, R.; Yao, S.; Zhai, P.; Li, S.; Yu, J.; Hou, Y.; Ma, D. Construction of Synergistic Fe5C2/Co Heterostructured Nanoparticles as an Enhanced Low Temperature Fischer-Tropsch Synthesis Catalyst. ACS Catal. 2017, 7, 5661–5667.
- Ismail, A.S.M.; Casavola, M.; Liu, B.; Gloter, A.; van Deelen, T.W.; Versluijs, M.; Meeldijk, J.D.; Stephan, O.; de Jong, K.P.; de Groot, F.M.F. Atomic-Scale Investigation of the Structural and Electronic Properties of Cobalt-Iron Bimetallic Fischer-Tropsch Catalysts. ACS Catal. 2019, 9, 7998–8011.
- Calderone, V.R.; Shiju, N.R.; Ferre, D.C.; Rothenberg, G. Bimetallic catalysts for the Fischer-Tropsch reaction. Green Chem. 2011, 13, 1950–1959.
- Zhang, J.; Abbas, M.; Chen, J. The Evolution of Fe Phases of a Fused Iron Catalyst during Reduction and Fischer-Tropsch Synthesis. Catal. Sci. Technol. 2017, 7, 3626–3636.
- Espinoza, R.L.; Steynberg, A.P.; Jager, B.; Vosloo, A.C. Low Temperature Fischer-Tropsch Synthesis from a Sasol Perspective. Appl. Catal. A Gen. 1999, 186, 13–26.
- de Smit, E.; Cinquini, F.; Beale, A.M.; Safonova, O.V.; van Beek, W.; Sautet, P.; Weckhuysen, B.M. Stability and Reactivity of ϵ-χ-θ Iron Carbide Catalyst Phases in Fischer-Tropsch Synthesis: Controlling μC. J. Am. Chem. Soc. 2010, 132, 14928–14941.
- Niemantsverdriet, J.W.; van der Kraan, A.M. Behavior of Metallic Iron Catalysts during Fischer-Tropsch Synthesis Studied with Mössbauer Spectroscopy, X-ray Diffraction, Carbon Content Determination, and Reaction Kinetic Measurements. J. Phys. Chem. B 1980, 84, 3363–3370.
- Chen, W.; Fan, Z.; Pan, X.; Bao, X. Effect of Confinement in Carbon Nanotubes on the Activity of Fischer-Tropsch Iron Catalyst. J. Am. Chem. Soc. 2008, 130, 9414–9419.
- Janbroers, S.; Louwen, J.N.; Zandbergen, H.W.; Kooyman, P.J. Insights into the Nature of Iron-based Fischer-Tropsch Catalysts from Quasi in situ TEM-EELS and XRD. J. Catal. 2009, 268, 235–242.
- Shroff, D.M.; Datye, A.K. The Importance of Passivation in the Study of Iron Fischer-Tropsch Catalysts. Catal. Lett. 1996, 37, 101–106.
- Moyer, M.M.; Karakaya, C.; Kee, R.J.; Trewyn, B. In Situ Formation of Metal Carbide Catalysts. ChemCatChem 2017, 9, 3090–3101.
- Zhang, Y.; Sirimanothan, N.; O’Brien, R.J.; Hamdeh, H.H.; Davis, B.H. Study of Deactivation of Iron-Based Fischer-Tropsch Synthesis Catalysts. Stud. Surf. Sci. Catal. 2001, 139, 125–132.
- Broos, R.J.P.; Zijlstra, B.; Filot, I.A.W.; Hensen, E.J.M. Quantum-Chemical DFT Study of Direct and H- and C-Assisted CO Dissociation on the χ-Fe5C2 Hägg Carbide. J. Phys. Chem. C 2018, 122, 9929–9938.
- Liu, X.; Cao, Z.; Zhao, S.; Gao, R.; Meng, Y.; Zhu, J.; Rogers, C.; Huo, C.; Yang, Y.; Li, Y.; et al. Iron Carbides in Fischer-Tropsch Synthesis: Theoretical and Experimental Understanding in Epsilon-Iron Carbide Phase Assignment. J. Phys. Chem. C 2017, 121, 21390–21396.
- Xu, K.; Sun, B.; Lin, J.; Wen, W.; Pei, Y.; Yan, S.; Qiao, M.; Zhang, X.; Zong, B. ε-Iron Carbide as a Low-Temperature Fischer-Tropsch Synthesis Catalyst. Nat. Commun. 2014, 5, 5783.
- Bukur, D.B.; Okabe, K.; Rosynek, M.P.; Li, C.; Wang, D.; Rao, K.R.P.M.; Huffman, G.P. Activation Studies with a Precipitated Iron Catalyst for Fischer-Tropsch Synthesis: I. Characterization Studies. J. Catal. 1995, 155, 353–365.
- Jung, H.; Thomson, W.J. Dynamic X-ray Diffraction Study of an Unsupported Iron Catalyst in Fischer-Tropsch Synthesis. J. Catal. 1992, 134, 654–667.
- Nagakura, S. Study of Metallic Carbides by Electron Diffraction Part III. Iron Carbides. J. Phys. Soc. Jpn. 1959, 14, 186–195.
- Wezendonk, T.A.; Santos, V.P.; Nasalevich, M.A.; Warringa, Q.S.E.; Dugulan, A.I.; Chojecki, A.; Koeken, A.C.J.; Ruitenbeek, M.; Meima, G.; Islam, H.-U.; et al. Elucidating the Nature of Fe Species during Pyrolysis of the Fe-BTC MOF into Highly Active and Stable Fischer-Tropsch Catalysts. ACS Catal. 2016, 6, 3236–3247.
- Königer, A.; Hammerl, C.; Zeitler, M.; Rauschenbach, B. Formation of Metastable Iron Carbide Phases after High-Fluence Carbon Ion Implantation into Iron at Low Temperatures. Phys. Rev. B Condens. Matter 1997, 55, 8143–8147.
- Herranz, T.; Rojas, S.; Perezalonso, F.; Ojeda, M.; Terreros, P.; Fierro, J. Genesis of Iron Carbides and Their Role in the Synthesis of Hydrocarbons from Synthesis Gas. J. Catal. 2006, 243, 199–211.
- de Smit, E.; Beale, A.M.; Nikitenko, S.; Weckhuysen, B.M. Local and Long Range Order in Promoted Iron-Based Fischer-Tropsch Catalysts: A Combined in situ X-ray Absorption Spectroscopy/Wide Angle X-ray. J. Catal. 2009, 262, 244–256.
- Galuszka, J.; Sano, T.; Sawicki, J.A. Study of Carbonaceous Deposits on Fischer-Tropsch Oxide-Supported Iron Catalysts. J. Catal. 1992, 136, 96–109.
- Hazemann, P.; Decottignies, D.; Maury, S.; Humbert, S.; Meunier, F.C.; Schuurman, Y. Selectivity Loss in Fischer-Tropsch Synthesis: The Effect of Carbon Deposition. J. Catal. 2021, 397, 1–12.
- Niu, L.; Liu, X.; Liu, J.; Liu, X.; Wen, X.; Yang, Y.; Xu, J.; Li, Y. Tuning Carburization Behaviors of Metallic Iron Catalysts with Potassium Promoter and CO/syngas/C2H4/C2H2 Gases. J. Catal. 2019, 371, 333–345.
- Chang, Q.; Zhang, C.; Liu, C.; Wei, Y.; Cheruvathur, A.V.; Dugulan, A.I.; Niemantsverdriet, J.W.; Liu, X.; He, Y.; Qing, M.; et al. Relationship between Iron Carbide Phases (ε-Fe2C, Fe7C3, and χ-Fe5C2) and Catalytic Performances of Fe/SiO2 Fischer-Tropsch Catalysts. ACS Catal. 2018, 8, 3304–3316.
- Chun, D.; Park, J.; Hong, S.; Lim, J.; Kim, C.; Lee, H.; Yang, J.; Hong, S.; Jung, H. Highly Selective Iron-based Fischer-Tropsch Catalysts Activated by CO2-containing Syngas. J. Catal. 2014, 317, 135–143.
- Lu, F.; Chen, X.; Lei, Z.; Wen, L.; Zhang, Y. Revealing the Activity of Different Iron Carbides for Fischer-Tropsch Synthesis. Appl. Catal. B 2021, 281, 119521.
- Wezendonk, T.A.; Sun, X.; Dugulan, A.I.; van Hoof, A.J.F.; Hensen, E.J.M.; Kapteijn, F.; Gascon, J. Controlled Formation of Iron Carbides and Their Performance in Fischer-Tropsch Synthesis. J. Catal. 2018, 362, 106–117.
- Zhao, H.; Liu, J.; Yang, C.; Yao, S.; Su, H.; Gao, Z.; Dong, M.; Wang, J.; Rykov, A.I.; Wang, J.; et al. Synthesis of Iron-Carbide Nanoparticles: Identification of the Active Phase and Mechanism of Fe-Based Fischer-Tropsch Synthesis. CCS Chem. 2021, 3, 2712–2724.
- Moodley, P.; Scheijen, F.J.E.; Niemantsverdriet, J.W.; Thüne, P.C. Iron Oxide Nanoparticles on Flat Oxidic Surfaces-Introducing a New Model Catalyst for Fischer-Tropsch Catalysis. Catal. Today 2010, 154, 142–148.
- Shipilin, M.; Degerman, D.; Lömker, P.; Goodwin, C.M.; Rodrigues, G.L.S.; Wagstaffe, M.; Gladh, J.; Wang, H.; Stierle, A.; Schlueter, C.; et al. In Situ Surface-Sensitive Investigation of Multiple Carbon Phases on Fe(110) in the Fischer-Tropsch Synthesis. ACS Catal. 2022, 12, 7609–7621.
- Lyu, S.; Wang, L.; Li, Z.; Yin, S.; Chen, J.; Zhang, Y.; Li, J.; Wang, Y. Stabilization of ε-iron Carbide as High-Temperature Catalyst under Realistic Fischer-Tropsch Synthesis Conditions. Nat. Commun. 2020, 11, 6219.
- Yang, C.; Zhao, H.; Hou, Y.; Ma, D. Fe5C2 Nanoparticles: A Facile Bromide-Induced Synthesis and as an Active Phase for Fischer-Tropsch Synthesis. J. Am. Chem. Soc. 2012, 134, 15814–15821.
More