The application of antibodies in cells was first shown in the early 1990s, and subsequently, the field of intracellular antibodies has expanded to encompass antibody fragments and their use in target validation and as engineered molecules that can be fused to moieties (referred to as warheads) to replace the Fc effector region of a whole immunoglobulin to elicit intracellular responses, such as cell death pathways or protein degradation. These various forms of intracellular antibodies have largely been used as research tools to investigate function within cells by perturbing protein activity. New applications of such molecules are on the horizon, namely their use as drugs per se and as templates for small-molecule drug discovery. The former is a potential new pharmacology that could harness the power and flexibility of molecular biology to generate new classes of drugs (herein referred to as macrodrugs when used in the context of disease control). Delivery of engineered intracellular antibodies, and other antigen-binding macromolecules formats, into cells to produce a therapeutic effect could be applied to any therapeutic area where regulation, degradation or other kinds of manipulation of target proteins can produce a therapeutic effect.
- intracellular antibodies
- macrodrugs
- domain antibodies
1. Introduction
2. Intracellular Antibody Fragments
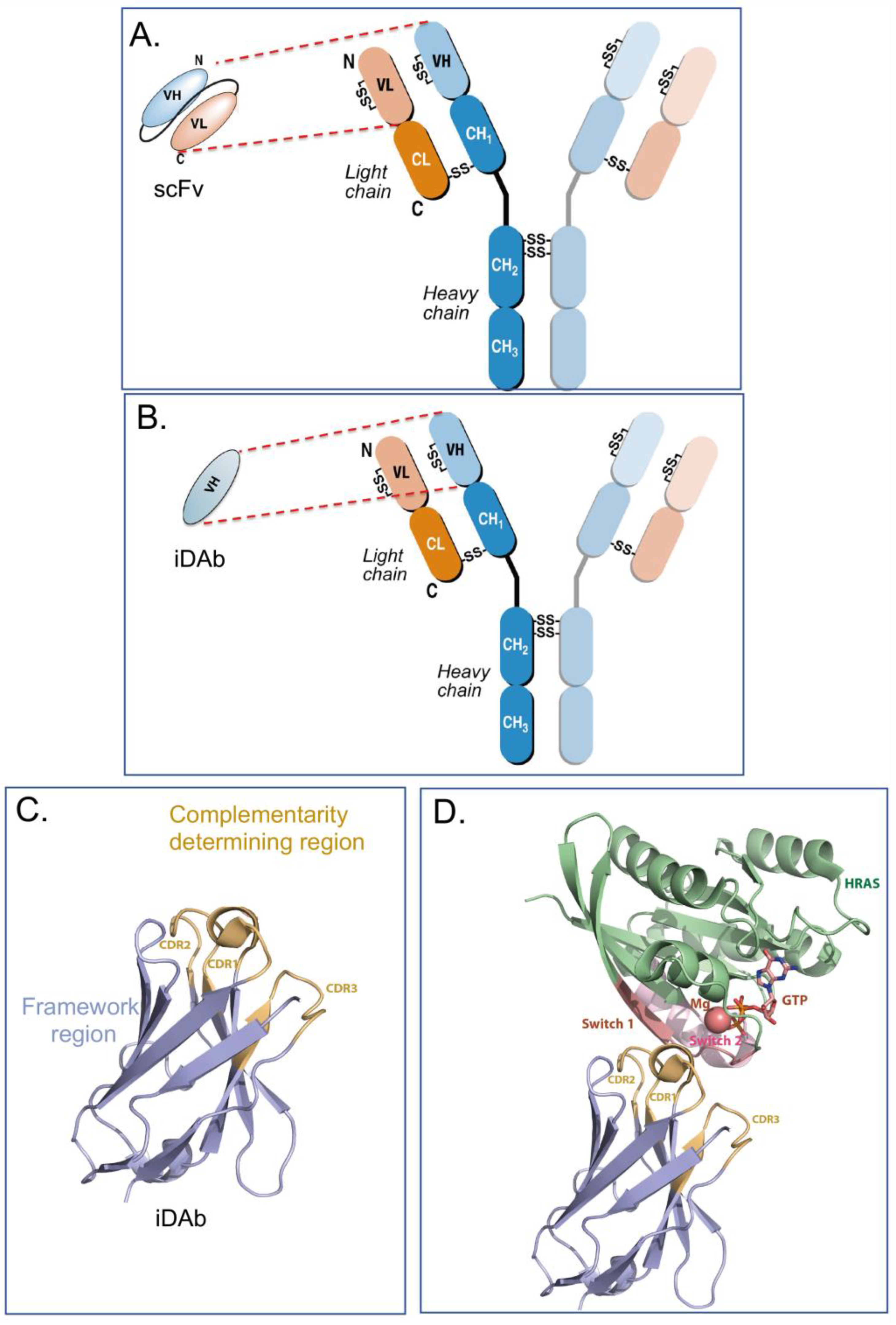
Collectively, human iDAbs or VHH nanobodies are a good choice for intracellular antibody use since they are single domains with one paratope comprising critical amino acids from three CDRs. This single paratope simplifies affinity manipulation if required [14][12], and does not involve linker segments such as those used between VH and VL in scFv format. In diverse scFv libraries, the association between VH and VL is often random to accommodate the hydrophobic interactions that naturally occur [15][13], indicating that many selected scFvs are only (or predominantly) binding through one of the variable region domains. For instance, an anti-RAS scFv was studied where only the VH showed detectable antigen binding [6]. Single-domain VH and VHH are effective antigen binders and thus lack constraints sometimes incurred by the presence of the VL. By sequencing many iDAbs isolated against a variety of antigens (including LMO2, RAS, CRAF, HOXA9, CMYC, TP53) a consensus framework VH sequence was obtained [7] (depicted in Figure 1C, illustrating the framework of the iDAb and the external CDR loops). The structure of an anti-RAS iDAb with HRAS (Figure 1D) formally validated mutant RAS PPI with signal transduction effector molecules as a cancer target [16][14].3. Specialising Intracellular Antibodies by Fusing them with Moieties to Affect Cell Phenotype or Viability
The ability of intracellular antibodies to produce a recognisable effect on target cells depends on a number of factors that will vary according to the function of the target protein. The effectiveness of an intracellular antibody is related to the half-life of antibody survival, which obviously reflects duration of contact with the antigen. Potent intracellular antibody inhibitors of protein–protein interaction (PPI) can be achieved, which allows for the blockade of a natural PPI since the Kd of the intracellular antibody can be made in the pM range, which is generally higher than that of the natural PPI. Occupancy is the major factor and slow koff facilitates the PPI inhibitor effect of intracellular antibodies via the prolongation of dimer interaction. Various other modifications in intracellular antibodies can be made that render the intracellular antibody more potent. These are referred to as warheads, (summarised in Figure 2), and simple protein engineering can derive new bivalent molecular structures that have the dual function of targeting an intracellular antigen and bringing it into the jurisdiction of an existing cellular process. Among the most potent of these highjacked mechanisms is the relocation of proteins within the cell by appending an endoplasmic reticulum (ER) signal peptide (KDEL, [19][15]), which locks up target antigens in the ER (Figure 2D). Alternatively, cytoplasmic proteins can be made into nuclear ones by the interacting intracellular antibodies, which have a nuclear localisation signal [20][11] (NLS, Figure 2C). Exploiting natural pathways within cells to induce a desired phenotype following binding of intracellular antibodies to their target is a powerful way to bring a target protein under the control of cellular elements not normally involved in this process. Two are illustrated in Figure 2. As a proof of concept, an anti-β-galactosidase scFv was directly fused to procaspase 3 (CP3) to form a dimer of dimers of the scFv-CP3 in contact with tetrameric b-galactosidase protein and inducing apoptosis as a result [21][16] (Figure 2E). This method (called antibody–antigen interaction-dependent apoptosis (AIDA)) was later performed using two separate iDAbs (one VH and one VL) that were cloned from an anti-RAS scFv [22][17]. The concept for AIDA technology was originally aimed to target fusion proteins that arise from commonly occurring chromosomal translocations, such as BCR-ABL fusion in Philadelphia-positive CML as an exemplar of this common type of tumour-associated protein [23][18]. Protein degradation was suggested as another route to control cellular phenotype by invoking proteosome degradation of targets in yeast [24][19]. This was followed by SIT technology in which the proteasome machinery was recruited for the targeted degradation of cellular proteins [25][20]. It was thus proposed that direct fusion of intracellular antibodies to ubiquitin ligases would cause specific degradation via the proteosome of the target protein after ubiquitination. Iterations of this approach have been developed in which direct fusion of various E3 ligases with intracellular antibody fragments (biodegraders) creates a binary complex in cells to lead to ubiquitination of target proteins and their proteosome degradation (Figure 2F).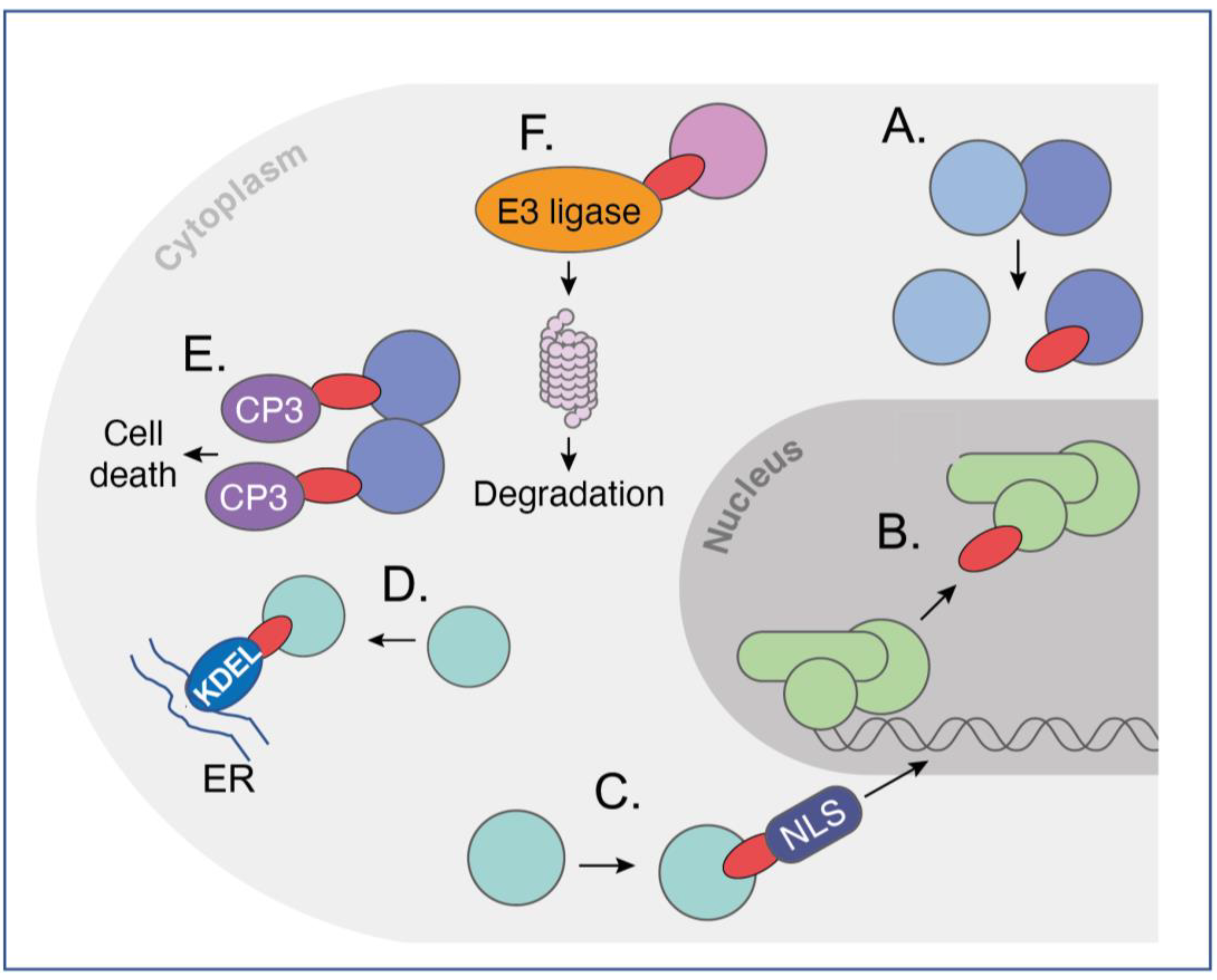
4. Options for Delivery of Intracellular Antibodies to Cells
5. Intracellular-Antibody-Derived Compounds: Bridging the Gap between Antibodies and Small Molecules
The difficulties in developing reliable and general delivery methods for using intracellular antibodies as macrodrugs per se prompted the development of Antibody-derived compound technology (Abd methodology) [51][28]. This was based on the observation that the iDAb CDR (the paratope) binding the antigen epitope has an approximately ten-amino-acid footprint and, based on molecular analysis [52][29], this would predict the small molecular equivalent of about 500 dA, just at the limit of the Lipinsky Rule of 5 which was an empirical evaluation of how drug-like a compound would be [53][30].
In the first-generation exposition of this Abd approach, a chemical fragment library was screened with HRAS protein, and chemical hits at the paratope–epitope interface were identified via competition with the intracellular antibody [51,54][28][31]. The key factor in this success was the pM Kd of the iDAb-RAS interaction (with low koff), which maintained the interaction of antigen–antibody during competition assessment in surface plasmon resonance. The intracellular antibody was in this case inhibiting compounds binding to the target.
Second-generation Abd was carried out on KRAS using a lower-affinity antibody fragment in order to find compounds that would directly inhibit the paratope–epitope interaction in the screen. As this requires low-affinity paratope–epitope interaction, a simple iDAb dematuration protocol was developed that depends only on the knowledge of the primary sequence of the iDAb [33][24] (i.e., no structural data are needed). These methods produced chemical matter that was pan-RAS since the iDAb involved binding to the RAS effector interaction region. The compounds were PPI inhibitors, as shown by cell-based BRET biosensors [55][32]. The third-generation Abd technology was designed to find compounds that displace intracellular antibodies that bind to disordered proteins, among which are many chromosomal translocation proteins and transcription factors. This disordered protein Abd screen used an anti-LMO2 intracellular antibody that had been used to confirm target validation in preclinical models [27][33]. It also implemented the dematured intracellular antibody approach but in a cell-based BRET screen, where the disordered protein was expressed in its normal cellular environment. The Abd technology is an antibody-based approach for drug discovery. It can not only be applied to intracellular antibodies but also to antibodies against the spectrum of diseases such as COVID-19, HIV and Ebola. An antibody binding to the membrane proximal external region (MPER) of the HIV-1 envelope has been used to guide the selection of small molecules that may be developed into therapeutic alternatives to the antibody [60][34]. The antibody-based approach to compound identification may be applicable to other clinical indications to replace antibodies where the cost of goods is very high and, without half-life extension properties, can be deleterious to patients due to the frequency of antibody treatment. Orally available chemical drugs are much more advantageous for patient use and compliance, as well as economy benefit, if specificity can be maintained while potency is increased.References
- Carlson, J.R. A new means of inducibly inactivating a cellular protein. Mol. Cell Biol. 1988, 8, 2638–2646.
- Biocca, S.; Neuberger, M.S.; Cattaneo, A. Expression and targeting of intracellular antibodies in mammalian cells. EMBO J. 1990, 9, 101–108.
- Marasco, W.A.; Haseltine, W.A.; Chen, S.Y. Design, intracellular expression, and activity of a human anti-human immunodeficiency virus type 1 gp120 single-chain antibody. Proc. Natl. Acad. Sci. USA 1993, 90, 7889–7893.
- Tanaka, T.; Rabbitts, T.H. Functional intracellular antibody fragments do not require invariant intra-domain disulfide bonds. J. Mol. Biol. 2008, 376, 749–757.
- Ward, E.S.; Gussow, D.; Griffiths, A.D.; Jones, P.T.; Winter, G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature 1989, 341, 544–546.
- Tanaka, T.; Lobato, M.N.; Rabbitts, T.H. Single domain intracellular antibodies: A minimal fragment for direct in vivo selection of antigen-specific intrabodies. J. Mol. Biol. 2003, 331, 1109–1120.
- Tanaka, T.; Rabbitts, T.H. Protocol for the selection of single-domain antibody fragments by third generation intracellular antibody capture. Nat. Protoc. 2010, 5, 67–92.
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448.
- Helma, J.; Cardoso, M.C.; Muyldermans, S.; Leonhardt, H. Nanobodies and recombinant binders in cell biology. J. Cell Biol. 2015, 209, 633–644.
- Roux, K.H.; Greenberg, A.S.; Greene, L.; Strelets, L.; Avila, D.; McKinney, E.C.; Flajnik, M.F. Structural analysis of the nurse shark (new) antigen receptor (NAR): Molecular convergence of NAR and unusual mammalian immunoglobulins. Proc. Natl. Acad. Sci. USA 1998, 95, 11804–11809.
- Tanaka, T.; Williams, R.L.; Rabbitts, T.H. Tumour prevention by a single antibody domain targeting the interaction of signal transduction proteins with RAS. EMBO J. 2007, 26, 3250–3259.
- Zeng, J.; Li, H.C.; Tanaka, T.; Rabbitts, T.H. Selection of human single domain antibodies recognizing the CMYC protein using enhanced intracellular antibody capture. J. Immunol. Methods 2015, 426, 140–143.
- Chothia, C.; Novotny, J.; Bruccoleri, R.; Karplus, M. Domain association in immunoglobulin molecules. The packing of variable domains. J. Mol. Biol. 1985, 186, 651–663.
- Tanaka, T.; Rabbitts, T.H. Interfering with RAS-effector protein interactions prevent RAS-dependent tumour initiation and causes stop-start control of cancer growth. Oncogene 2010, 29, 6064–6070.
- Hammond, C.; Helenius, A. Quality control in the secretory pathway: Retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J. Cell Biol. 1994, 126, 41–52.
- Tse, E.; Rabbitts, T.H. Intracellular antibody-caspase-mediated cell killing: An approach for application in cancer therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 12266–12271.
- Chambers, J.S.; Brend, T.; Rabbitts, T.H. Cancer cell killing by target antigen engagement with engineered complementary intracellular antibody single domains fused to pro-caspase3. Sci. Rep. 2019, 9, 8553.
- Tse, E.; Lobato, M.N.; Forster, A.; Tanaka, T.; Chung, G.T.; Rabbitts, T.H. Intracellular antibody capture technology: Application to selection of intracellular antibodies recognising the BCR-ABL oncogenic protein. J. Mol. Biol. 2002, 317, 85–94.
- Zhou, P.; Bogacki, R.; McReynolds, L.; Howley, P.M. Harnessing the ubiquitination machinery to target the degradation of specific cellular proteins. Mol. Cell 2000, 6, 751–756.
- Melchionna, T.; Cattaneo, A. A protein silencing switch by ligand-induced proteasome-targeting intrabodies. J. Mol. Biol. 2007, 374, 641–654.
- Lobato, M.N.; Rabbitts, T.H. Intracellular antibodies and challenges facing their use as therapeutic agents. Trends Mol. Med. 2003, 9, 390–396.
- Stumpp, M.T.; Dawson, K.M.; Binz, H.K. Beyond Antibodies: The DARPin((R)) Drug Platform. BioDrugs 2020, 34, 423–433.
- Sha, F.; Salzman, G.; Gupta, A.; Koide, S. Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 2017, 26, 910–924.
- Tanaka, T.; Thomas, J.; Van Montfort, R.; Miller, A.; Rabbitts, T. Pan RAS-binding compounds selected from a chemical library by inhibiting interaction between RAS and a reduced affinity intracellular antibody. Sci. Rep. 2021, 11, 1712.
- Bery, N.; Bataille, C.J.R.; Russell, A.; Hayes, A.; Raynaud, F.; Milhas, S.; Anand, S.; Tulmin, H.; Miller, A.; Rabbitts, T.H. A cell-based screening method using an intracellular antibody for discovering small molecules targeting the translocation protein LMO2. Sci. Adv. 2021, 7, eabg1950.
- Francis, A.I.; Ghany, S.; Gilkes, T.; Umakanthan, S. Review of COVID-19 vaccine subtypes, efficacy and geographical distributions. Postgrad Med. J. 2022, 98, 389–394.
- Town, J.; Pais, H.; Harrison, S.; Stead, L.F.; Bataille, C.; Bunjobpol, W.; Zhang, J.; Rabbitts, T.H. Exploring the surfaceome of Ewing sarcoma identifies a new and unique therapeutic target. Proc. Natl. Acad. Sci. USA 2016, 113, 3603–3608.
- Quevedo, C.E.; Cruz-Migoni, A.; Bery, N.; Miller, A.; Tanaka, T.; Petch, D.; Bataille, C.J.R.; Lee, L.Y.W.; Fallon, P.S.; Tulmin, H.; et al. Small molecule inhibitors of RAS-effector protein interactions derived using an intracellular antibody fragment. Nat. Commun. 2018, 9, 3169.
- Janin, J.; Chothia, C. The structure of protein-protein recognition sites. J. Biol. Chem. 1990, 265, 16027–16030.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26.
- Cruz-Migoni, A.; Canning, P.; Quevedo, C.E.; Bataille, C.J.R.; Bery, N.; Miller, A.; Russell, A.J.; Phillips, S.E.V.; Carr, S.B.; Rabbitts, T.H. Structure-based development of new RAS-effector inhibitors from a combination of active and inactive RAS-binding compounds. Proc. Natl. Acad. Sci. USA 2019, 116, 2545–2550.
- Bery, N.; Cruz-Migoni, A.; Bataille, C.J.; Quevedo, C.E.; Tulmin, H.; Miller, A.; Russell, A.; Phillips, S.E.; Carr, S.B.; Rabbitts, T.H. BRET-based RAS biosensors that show a novel small molecule is an inhibitor of RAS-effector protein-protein interactions. Elife 2018, 7, 37122.
- Tanaka, T.; Sewell, H.; Waters, S.; Phillips, S.E.; Rabbitts, T.H. Single domain intracellular antibodies from diverse libraries: Emphasizing dual functions of LMO2 protein interactions using a single VH domain. J. Biol. Chem. 2011, 286, 3707–3716.
- Xiao, T.; Frey, G.; Fu, Q.; Lavine, C.L.; Scott, D.A.; Seaman, M.S.; Chou, J.J.; Chen, B. HIV-1 fusion inhibitors targeting the membrane-proximal external region of Env spikes. Nat. Chem. Biol. 2020, 16, 529–537.