Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 3 by Jessie Wu.
Alkenes and their related analogs are a class of ideal starting materials for the construction of complex molecules, because they are readily available in bulk quantities from renewable resources and petrochemical feedstocks. They are also considered to be the most cost-effective and widely used raw material for organic synthesis, and due to the diversity of functional groups, they are used very frequently in different chemical industries. As basic functionalities, the exploration of efficient methods for the selective functionalization of alkenes has been a continuous pursuit throughout the history of organic chemistry.
- photoredox-catalysis
- difunctionalization of alkenes
- electron donor-acceptor complexes
- pyridinium
- radical
1. 1,2-Dicarbofunctionalization of Alkenes
In 2019, Glorius and co-workers [1] disclosed an elegant method of the 1,2-dicarbofunctionalization of aryl alkenes with Katritzky pyridinium salts as the radical precursors and electron-rich indoles as the coupling partners. With a catalytic amount of [Ir(dtbbpy)(ppy)2](PF6), a number of densely functionalized 1,1-diarylalkanes could be obtained in 25–95% yields (Scheme 1). Notably, pyridine-containing products, thioether-functionalized products, ester-containing products, Boc-protected amino-functionalized products and all-carbon quarternary centers are successfully produced in this system. Dipeptide-derived Katritzky pyridinium salt is also amenable in this transformation. Further, mechanistic studies suggest that the reaction began with the reduction of Katritzky salt by excited photocatalyst to afford the corresponding radical, which was captured by the aryl alkenes to give the arylmethyl radical intermediate. This intermediate could be oxidized to produce an arylmethyl carbocation intermediate, which was trapped by the nucleophilic aromatic compound to furnish the target product.
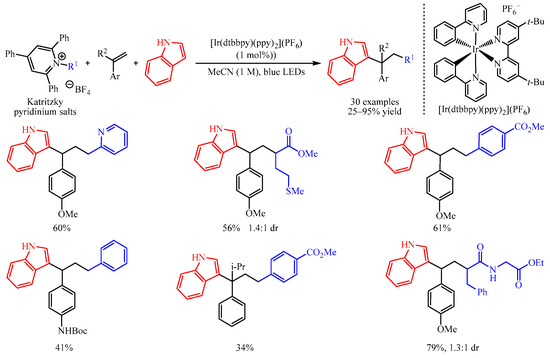
Scheme 1. Dicarbofunctionalization of aryl alkenes with Katritzky pyridinium salts.
Subsequently, the Molander group [2] adopted another kind of radical progenitor alkyl N-(acyloxy)phthalimide ester to trigger the 1,2-dicarbofunctionalization of electron-rich aryl and heteraryl substituted olefins with organotrifluoroborates as the carbon-centered nucleophiles (Scheme 2). This protocol provides a robust implement for the carbo-alkynylation, -arylation, -allylation, or -alkenylation of alkenes by simply altering the structure of organotrifluoroborates. The author suggested that this reaction proceeds through photochemical radical/polar oxidation to afford a key carbocation species 1 with the extrusion of carbon dioxide and Phth from alkyl N-(acyloxy)phthalimide esters. This carbocation species then underwent subsequent coupling with organoboron nucleophiles to produce the desired product. Notably, the strained cyclobutane subunit, the Boc-protected amine, halogens, heterocyclic moiety, the bridged bicycle and acyclic moieties were all tolerated in this reaction. Furthermore, a substrate derived from estrone was also viable and delivering the corresponding steroid derivative in good yield [3].
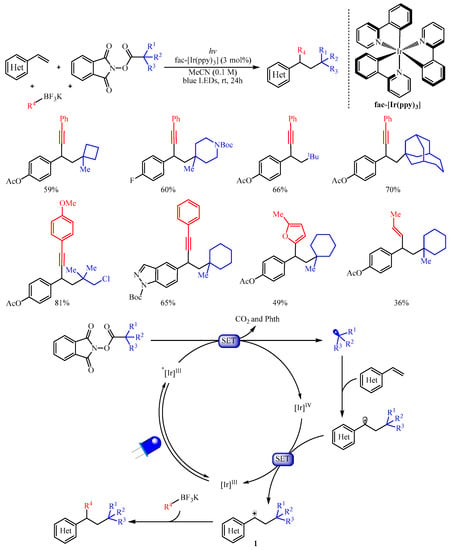
Scheme 2. Dicarbofunctionalization of alkenes with alkyl N-(acyloxy)phthalimide esters.
Recently, Huang and co-workers [4] addressed the (Ir[dF(CF3)ppy]2(dtbpy))PF6 catalyzed difunctionalization of alkenyl ketones for the synthesis of tetralones and cyclopropane compounds with α-carbonyl alkyl bromide as the radical source (Scheme 3). With the irradiation of visible light, the Ir complex could promote the reduction of α-carbonyl alkyl bromides to deliver the corresponding radical species. Subsequently, this radical was trapped by the alkenyl group of the substrate to produce the secondary alkyl radical intermediate, which preceded a further cyclization process to obtain the product. Interestingly, with gem-dimethyl substituted alkenes as the substrate, intramolecular cyclization occurred between the aryl group and secondary alkyl radical section, followed by oxidization with the Ir-complex to afford the six-membered cyclic product tetralones. However, when β,γ-unsaturated arylketones were introduced, the cyclopropane derivatives could be achieved through the loss of a proton and intramolecular three-membered cyclization.
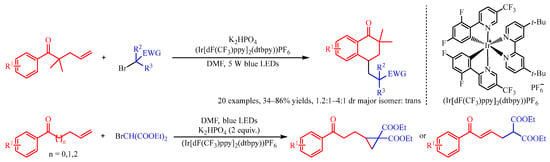
Scheme 3. Iridium-photocatalyzed cyclization of alkenyl ketones with α-carbonyl alkyl bromides.
In 2022, Mao and co-workers [5] developed another radical progenitor [Ph3PCF2H]+Br−triggered, visible light-mediated tandem difluoromethylation/cyclization reaction with alkenyl aldehydes as the starting material (Scheme 4). By utilizing fac-Ir(ppy)3 as the catalyst, a range of CF2H-substituted chroman-4-one skeletons could be achieved in moderate to good yields, and good chemoselectivity under mild reaction conditions. In this transformation, the fac-Ir(ppy)3 could be activated to generate the excited-state fac-IrIII(ppy)3* under irradiation with a 5 W blue LED, which then reduced [Ph3PCF2H]+Br− to produce the vital •CF2H radical through a single-electron transfer (SET) process to promote the further intramolecular cyclization. In addition, substrates derived from phenolphthalein and coumarin were also viable, generating the corresponding products in good yields.
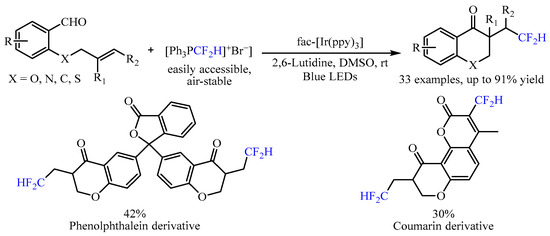
Scheme 4. [Ph3PCF2H]+Br−-triggered difluoromethylation/cyclization of alkenyl aldehydes.
With the same catalyst, Dolbier and co-workers [6] realized the photoredox-catalyzed addition of difluoromethyl radical to trimethylsilyloxy-substituted alkenes, allowing the construction of a diverse series of difluoromethyl ketones via neophyl-like aryl and heteroaryl migrations (Scheme 5). With the irradiation of 390–780 nm LEDs, both 1,2- and 1,4-functional group migrations could be achieved, delivering the corresponding product with 32–96% yields. The reaction was initiated by the photoexcited fac-Ir(ppy)3 catalyst to reduce CF2HSO2Cl via SET with the release of chloride and SO2, generating the vital •CF2H radical. This radical was then captured by the alkenyl group and further underwent aryl ipso-migration to deliver an arylmethyl radical, which was subsequently oxidized by the high-valent Ir catalyst and underwent the loss of the trimethylsilyl group to furnish the final product.
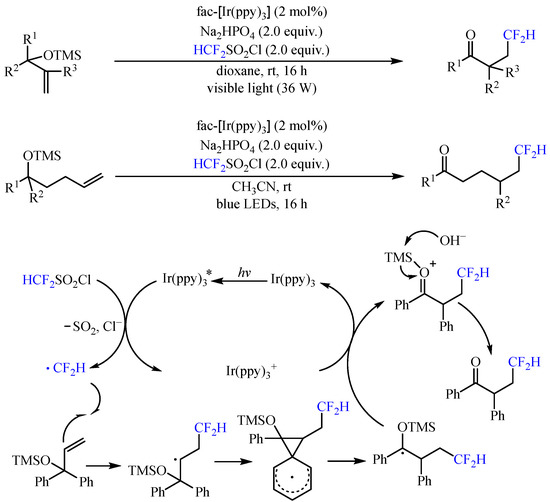
Scheme 5. Photoredox-catalyzed addition of difluoromethyl radical to trimethylsilyloxy-substituted alkenes.
The CF3 group is one of the most important functional groups for drug research, and numerous efforts have been devoted to the incorporation of this group into specific molecules [7]. In this regard, Kim et al. [8] developed a (Ir[dF(CF3)ppy]2(dtbpy))PF6-catalzyed functionalization of 2-vinylphenols with 1,3-diketones as the coupling partner and the famous Umemoto’s reagent as the trifluoromethylating reagent (Scheme 6). This reaction resulted in the capture of in situ-generated trifluoromethylated ortho-quinone methides from 2-vinyl phenols, followed by conjugate addition and deacetylation. With 2,2,2-trifluoroethanol (TFE) as the solvent, a variety of γ-trifluoromethylated ketones could be generated in 51–92% yields under the irradiation of 5 W blue LEDs (λmax = 455 nm). In addition, the trifluoromethylated ketone products could be successfully cyclized to 4-(2,2,2-trifluoroethyl)-4H-chromenes in good yields with an equivalent amount of Sc(OTf)3.
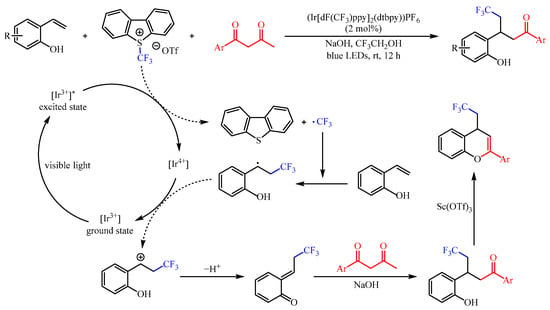
Scheme 6. Photoredox-catalzyed functionalization of 2-vinylphenols with 1,3-diketones.
2. Carbon-Hetero Bond Formation
In 2022, Chen’s group [9] developed a visible light-induced fac-Ir(ppy)3, catalyzed three-component Ritter reaction with alkenes, nitriles, and α-bromo nitriles/esters as the substrates, delivering various structurally diverse γ-amino nitriles/acids (Scheme 7). The authors disclosed that the acetonitrile radical or ester group-containing radical could be generated through the metalla-photoredox catalyst fac-Ir(ppy)3-promoted SET process from α-bromo nitriles or α-bromoesters, respectively. Importantly, the introduction of KF is critical for the generation of the acetimidoyl fluoride intermediate, which undergoes the Ritter reaction, thus delivering the amino-alkylation product.
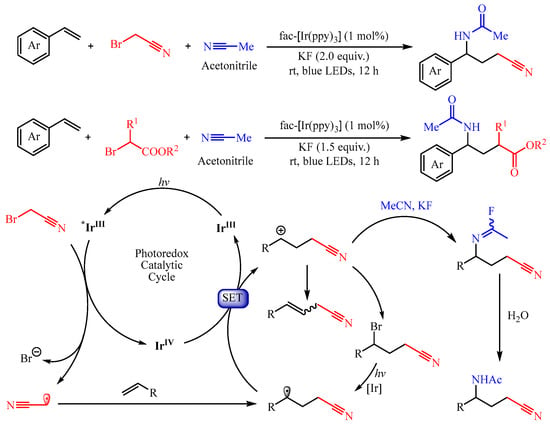
Scheme 7. Three-component aminofluorination and aminochlorination of olefins.
Molander’s group [10] reported the amidoarylation of the amide group containing alkenes by merging photoredox proton-coupled electron transfer (PCET) with nickel catalysis for the synthesis of five-membered heterocyclics with aryl halides (Scheme 8). In this protocol, the [Ir-photocatalyst] (3 mol%) was utilized as the photocatalyst and Ni-(dMeObpy)(H2O)2Br2 (6 mol%) was the catalyst for the radical-mediated coupling process. This strategy has granted access to an array of complicated molecules bearing a pyrrolidinone core from readily available alkenyl amides and aryl- or heteroaryl-halides. Additionally, this transformation is not limited to amides, as carbamates and ureas were also viable. Disappointingly, the reaction failed for substrates bearing protic functional groups. Mechanistic studies, including hydrogen-bond affinity constants, control experiments and cyclic voltammetry studies, have confirmed that the reaction followed a proton-coupled electron transfer (PCET) procedure. Initially, the formation of an amidyl radical 2 via PCET occurred, which was followed by fast 5-exo-trig cyclization to generate the alkyl radical 3. Subsequently, combination with the nickel catalyst formed a Ni(I)-complex 4, which underwent oxidative addition with aryl halides, resulting in the Ni-intermediate 5. The reductive elimination of 5 generated the desired product 6 and the Ni(I)-halide complex 7 to facilitate the next catalytic cycle.
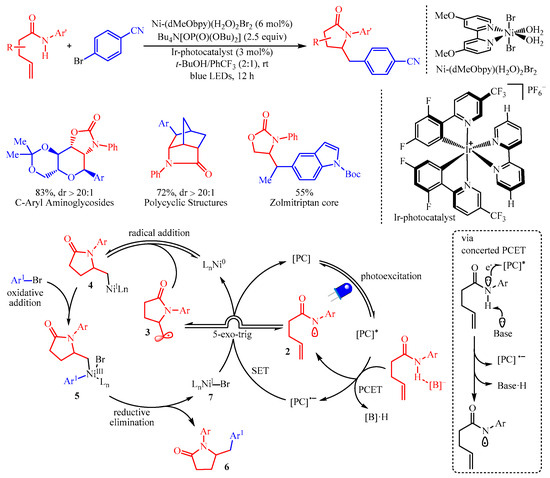
Scheme 8. The amidoarylation of alkenes with aryl halides via photoredox PCET/Ni-catalysis.
Bromodifluoromethanephosphonates represents another kind of difluoromethylation reagent, and the use of such reagents could not only introduce the CF2 group, but also introduced a phosphate functional group [11]. The photocatalyzed intermolecular aminodifluoromethylphosphonation of aryl alkenes was successfully developed by Yang’s group [12] for the synthesis of α,α-difluoro-γ-aminophosphonates (Scheme 9). In this protocol, fac-[Ir(ppy)3] was employed as the photocatalyst and arylamines as the nucleophilic reagent. The diethyl or diisopropyl bromodifluoromethylphosphate was tolerated in this transformation; however, (bromodifluoromethyl)diphenylphosphine oxide failed to give the desired product. Moreover, this reaction could be applied to the modification of complex chiral arylamines, such as (R)-BINAM ((R)-(+)-2,2′-diamino-1,1′-binaphthalene).
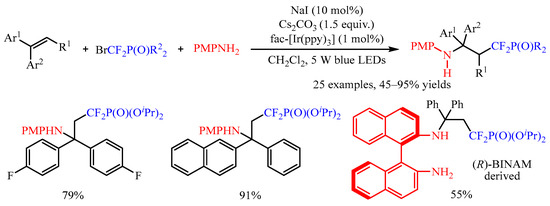
Scheme 9. Photocatalyzed aminodifluoromethylphosphonation of alkenes.
α-Difluoroacetates is a class of efficient difluoroacetylation reagent, and it could also be introduced to the difunctionalization of alkenes [13]. In 2022, Xi’s group [14] developed the photoredox-catalyzed hydroxydifluoroacetylation of aryl alkenes with FSO2CF2CO2Me and H2O as the coupling partners (Scheme 10). The -CF2CO2Me and -OH groups could be simultaneously introduced into the molecules with fac-[Ir(ppy)3] as the photocatalyst in good to high yields with high regioselectivity. Interestingly, when disubstituted alkenes were used in the reaction, the difluorolactone compound was only isolated after a general work-up due to the congestion effect of the substituents. Subsequently, the same group disclosed the fluorodifluoroacetylation of aromatic alkenes with this difluoroacetylation reagent. In this reaction, the Et3N·3HF was introduced as the nucleophilic reagent, and an array of γ-functionalized difluoroacetates could be obtained with 60–84% yields [15].
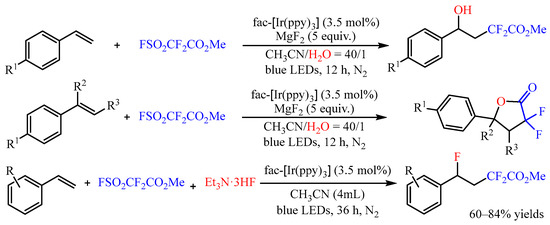
Scheme 10. Difunctionalization of alkenes with FSO2CF2CO2Me.
In 2020, Shen’s group [16] developed a reactive electrophilic selenium ylide-based trifluoromethylating reagent and realized the difunctionalization of aryl alkenes using an array of nucleophiles, such as amines, azides, alcohols, water, as well as electron-rich arenes. The Lewis acid Sc(OTf)3 and photoredox catalyst fac-Ir(ppy)3 were selected for use in the synergistic catalyst system to promote the 1,2-difunctionalization of olefins, providing a unified route to functionalized trifluoromethylated compounds with good to excellent yields (Scheme 11). Mechanistic studies have demonstrated that the Lewis acid Sc(OTf)3 could activate this trifluoromethylating process via generating the complex Sc(OTf)3•3(8) through Lewis acid–Lewis base interaction. It is worth mentioning that the radical addition/cyclization sequence occurred smoothly when tosyl-protected allylamine was used as the substrate [17].
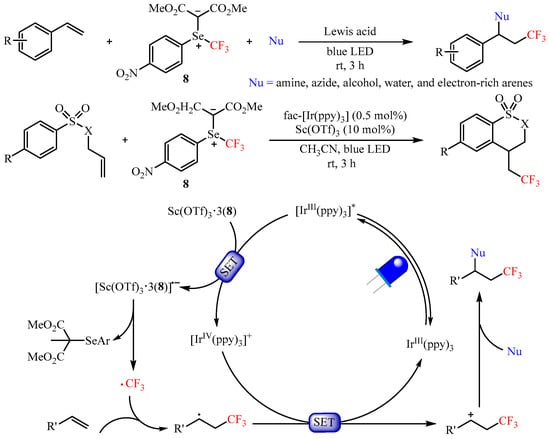
Scheme 11. Difunctionalization of aryl alkenes with selenium ylide-based trifluoromethylating reagent.
Recently, trifluoromethylthiomoiety (SCF3) has attracted increasing attention due to its high Hansch’s hydrophobicity parameter, and numerous methods for its preparation have been described in the literature [18]. As regards the difunctionalization of olefins, Magnier and co-workers [19] accomplished the carbotrifluoromethylthiolation of acrylamides or aryl alkenes with N-trifluoromethylthiosaccharin as the source of the SCF3 radical (Scheme 12). With fac-[Ir(ppy)3] as the photocatalyst, a series of N-aryl acrylamides andstyrenes could be difunctionalized in both an intramolecular and an intermolecular manner, providing the corresponding products in good to excellent yields. Additionally, the author demonstrated that the vital •SCF3 radical was generated by the photoexcited Ir-catalyzed reduction of N-trifluoromethylthiosaccharin through spin trapping/electron paramagnetic resonance (ST/EPR) experiments.
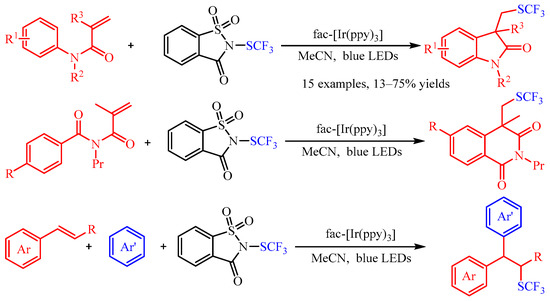
Scheme 12. Carbotrifluoromethylthiolation of acrylamides or aryl alkenes with N-trifluoromethylthiosaccharin.
In 2021, Zhang and co-workers [20] developed a synergistic photoredox and Cu(II)-catalyzed fluoroalkylphosphorothiolation of inactivated alkenes via a radical process. In this catalysis, the fluoroalkyl halides (RfX) was introduced as the radical precursor, and the phosphorus compound P(O)SH or P(S)SH was selected as the coupling partner (Scheme 13). A wide range of S-alkyl phosphorothioates and phosphorodithioates bearing β-monofluoroalkyl, -difluoroalkyl, -trifluoromethyl, or -perfluoroalkyl substitutes could be easily fabricated with good yields and moderated to good diastereoselectivity. Importantly, this transformation could be applied to the late-stage functionalization of bioactive molecules [21]. Through mechanistic studies, the authors proposed a possible mechanism. Firstly, the catalyst IrIII could be excited to generate the IrIII* with blue light irradiation, and this highly active species reduces BrCF2COOEt to produce an ethyl difluoroacetate radical through the SET process. Then, the in situ-formed ethyl difluoroacetate radical is captured by the C=C bond of styrene, generating the alkyl radical intermediate. Simultaneously, the phosphorothiolation compound (EtO)2P(O)SH reacts with the Cu(II)-catalyst, yielding the corresponding active (EtO)2P(O)S-Cu(I) intermediate, which further produces a (EtO)2P(O)S-Cu(II) complex 9 via oxidation with the high-valent Ir(IV) species, releasing the iridium (Ir) to finish the photo-redox cycle. This complex 9 can cooperate with alkyl radical 10 to produce a Cu(III) species 11, which undergoes reductive elimination to regenerate the Cu(I) species to close the Cu-catalysis cycle.
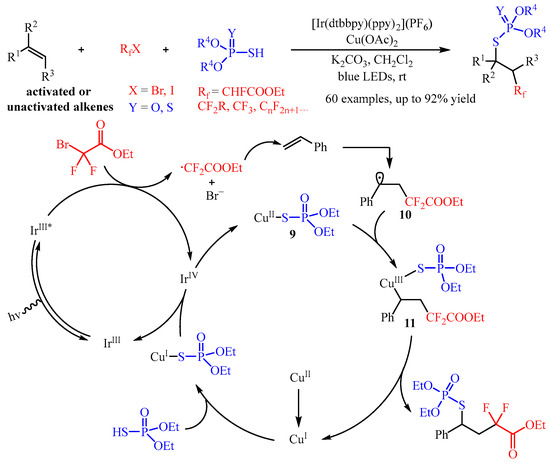
Scheme 13. Synergistic photoredox and copper-catalyzed fluoroalkylphosphorothiolation of alkenes.
In the same year, Glorius’ group [22] developed an elegant [Ir(dtbbpy)(ppy)2](PF6)-catalyzed difunctionalization of alkenes via a base-controlled reincorporation/release of SO2 strategy (Scheme 14). With this chemo-divergent strategy, various valuable γ-trifluoromethylated ketones and trifluoromethylated sulfonyl ketones could be constructed from the same starting materials. This catalysis exhibited high chemoselectivity with a broad substrate scope. Simple alkenes, such as propene, ethylene, 1-hexene, decene and allylcyclohexane, were amenable. Multi-substituted alkenes, including 1,2-disubstituted, α-methyl substituted, trisubstituted, and tetrasubstituted alkenes, were proved as suitable substrates. Various functional groups including ester, ether, ketone, succinimide, phosphate ester, and polyfluoroalkyl groups are well tolerated. Notably, this protocol could be scaled up and applied in the construction of complicated molecules. Furthermore, one-pot synthesis with alkenes, ketones, and sulfonic anhydride as the starting materials was also viable. Through systematic mechanistic studies, the author proposed a radical chain mechanism for this transformation. Firstly, the •CF3 radical was generated from the enol triflate substrate via SET catalysis, which was followed by the attack of alkenes to produce a carbon-centered radical. Then, this radical underwent two different transformations with the presence of different bases. In catalytic cycle A, intermediate 12 can react with enol triflate and then undergo further fragmentation to finish the desired product by the release of the SO2. In this process, the excess KOH could consume the generated SO2 and facilitate this transformation. However, when introducing K2CO3 as the base, the intermediate K2S2O could be generated and react with 12 to deliver 13, which could add to the enol triflate substrate to selectively produce the trifluoromethylated sulfonyl ketones [23].
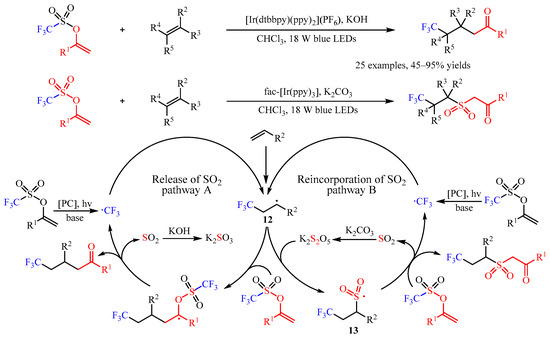
Scheme 14. Base-controlled chemo-divergent difunctionalization of alkenes.
Fluoroalkanesulfinate salts ((RFSO2)nM), such as the Langlois reagent CF3SO2Na, could also be utilized as dual fluoroalkyl (RF) and sulfur dioxide (SO2) sources by the action of photoredox catalysis [24][25]. In 2020, Akita and co-workers [26] found that the Ir-complex [Ir{dF(CF3)ppy}2(bpy)](PF6) could promote the photoredox catalyzed fluoroalkyl-sulfonylation of alkenes with CF3SO2Na, (CF2HSO2)2Zn, MeCF2SO2Na, and 4-BrC6H4CH2CF2SO2Na as the radical precursors. Under visible light irradiation (425 nm), the reaction of fluoroalkanesulfinate salts with alkenes generated a trifluoromethyl-sulfonylated product, which could be transformed to a more stable methanesulfonylated product via the treatment of the reaction mixture with MeI (Scheme 15). Besides substituted styrenes, other alkenes including aliphatic alkenes, allylamine derivatives and pyridyl substituted alkenes were compatible with this present reaction, delivering the corresponding products in 23–69% yields.
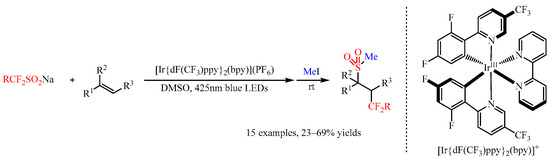
Scheme 15. Photocatalyzed fluoroalkyl-sulfonylation of alkenes with Langlois reagent.
Shortly afterwards, Yi and co-workers [27] found that the same catalyst promoted the trifluoromethyl-thiotrifluoromethylation of alkenes by using CF3SO2Na as both CF3 and SCF3 source (Scheme 16). This photocatalysis strategy provided a facile method to a variety of vicinal trifluoromethylthio-trifluoromethylated compounds with 31–91% yields. In addition, an array of mechanistic investigations demonstrated that the reaction occurred through a photocatalyzed dual-oxidative process. Firstly, a long-lived triplet excited-state species IrIII* was generated through the SET pathway. Then, another SET between such species and CF3SO2Na resulted in the trifluoromethyl radical •CF3 and IrII species. The in situ-formed •CF3 radical then reacted with alkenes to generate a carbon-centered radical intermediate 14. Meanwhile, CuSCF3 could be generated in situ from CuI and CF3SO2Na with the assistance of PPh3, which was followed by oxidation to deliver a CuII species via SET. Subsequently, the intermediate 14 underwent single-electron oxidative addition to CuII-SCF3 to form species 15, a formally CuIII intermediate. Lastly, reductive elimination from intermediate 15 would yield the desired product by regenerating the corresponding CuI species.
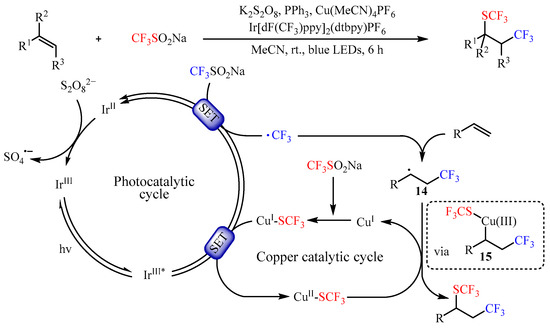
Scheme 16. Photocatalyzed trifluoromethylthiotrifluoromethylation of alkenes using CF3SO2Na.
Recently, the N-Ts-protected 1-aminopyridine salt was developed as a N-radical progenitor in photoredox catalytic reactions [28]. The subjection of this compound to the mixture of olefins with a suitable nucleophilic reactant provided a chance for the construction of olefin-difunctionalized products. In this regard, Xu and co-workers [29] disclosed the Ir-catalyzed three-component aminofluorination or aminochlorination of styrenes with this shelf-stable nitrogen-radical precursor, and hydrogen fluoride-pyridine (py•9HF) or pyridine hydrochloride (py•HCl) as the nucleophilic halogen source. An array of styrenes were converted to the corresponding fluorosulfonamides or chlorosulfonamides in yields ranging from 34% to 86%, with the diastereoselectivities ranging from 1.2:1 to 4:1 in favor of the trans diastereomers (Scheme 17).
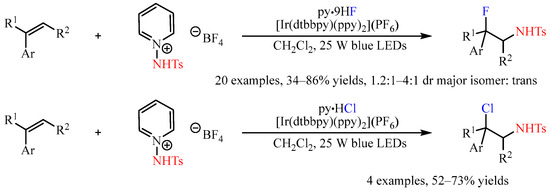
Scheme 17. Three-component aminofluorination and aminochlorination of olefins with N-Ts-protected 1-aminopyridine salt.
With Umemoto’s reagent as the trifluoromethylating reagent, Akita and co-workers [30] accomplished the three-component intermolecular oxytrifluoromethylation of alkenes with oxygenic nucleophiles. The introduction of a CF3 group and various oxygenic functional groups, including hydroxy, alkoxy, as well as carboxy groups, to C=C bonds has been realized by employing fac-Ir(ppy)3 as the photoredox catalysis under irradiation with blue LEDs or sunlight (Scheme 18) [31]. Notably, this reaction could be applied to styrenes with halogen atoms or ester groups, such as AcO and Bpin. For internal alkenes, the reaction occurred smoothly, with the diastereoselectivity ranging from 1:1 to 1:10. Notably, the electron-rich alkene 3,4-dihydro-2H-pyran also provided a suitable substrate in this photocatalytic system.
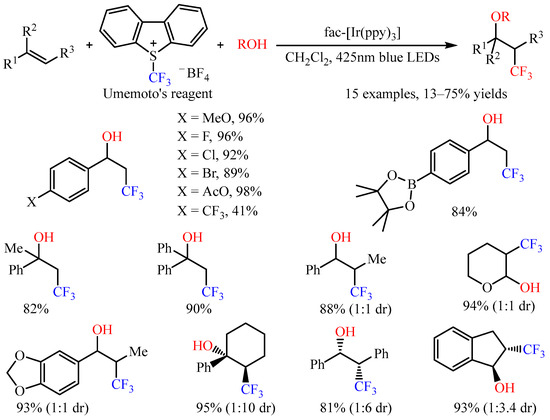
Scheme 18. Photocatalyzed oxytrifluoromethylation of alkenes with Umemoto’s reagent.
Xiao and co-workers [32] found that the exposure of 2-vinylphenols to the mixture of Umemoto’s reagent and sulfur ylides under photoredox catalysis with fac-Ir(4′-CF3ppy)3 enabled a multicomponent cyclization reaction, providing access to trifluoromethylated 2,3-dihydrobenzofurans (Scheme 19). The substrate scope of sulfur ylides proved to be broad, and substrates bearing furan- and thiophene- substituents were amenable. Importantly, the sulfonium bromide could be used directly, producing the desired product with good yield. The reaction with 2-vinyl phenols bearing several functional groups such as halogen and nitro groups worked well to furnish the vinyl difunctionalized products. Interestingly, the cyanoalkylated 2,3-dihydrobenzofuran could be obtained in a 70% yield, when using iodoacetonitrile instead of Umemoto’s reagent as the radical source and Ru(bpy)3Cl2•6H2O as the photocatalyst. Notably, the use of (R)-BINOL-derived sulfur ylide under standard conditions resulted in the corresponding product in an 18% yield with 25% ee. On the basis of the mechanistic studies, the author proposed a plausible mechanism. First, the relatively stable benzyl radical was generated through the SET process, which was followed by further oxidization by the oxidizing state IrIV via the SET process, forming a benzylic cation. Subsequently, this benzylic cation intermediate underwent deprotonation to produce a reactive o-QM intermediate, which participated in a formal [4+1] annulation with the sulfur ylide to obtain the desired product.
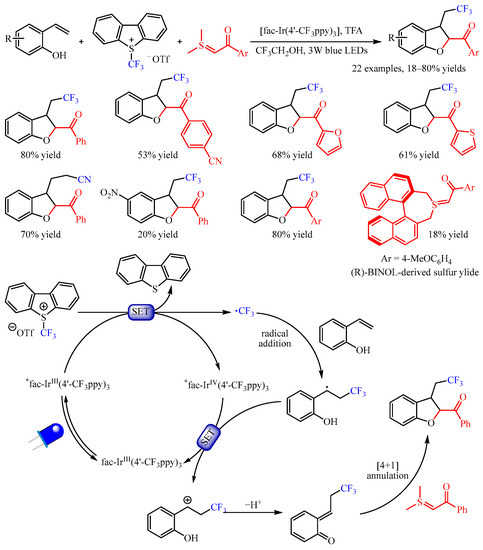
Scheme 19. Photoredox-catalyzed difunctionalization of alkenes through the generation of o-quinone methides.
Subsequently, the same group found that a light-drivenα-carbonyl alkyl radical could be formed via the SET-reduction of sulfur ylides, and this process can be coupled with the difunctionalization of aryl alkenes under the Ir-catalyzed photoredox reaction [33]. Notably, this transformation presented a broad substrate scope with high functional group tolerance for both sulfur ylides and alkenes, affording a practical method to achieve structurally diverse γ-hydroxy carbonyl compounds with up to 99% yield. The authors also proposed a possible mechanism, as outlined in Scheme 20. Initially, the sulfur ylide 16 could undergo the reaction with 2,2,2-trifluoroethanol and Et3N•3HF to produce the corresponding sulfonium salt, which was transformed to the α-carbonyl carbon radical via SET-reduction with the irradiation of the LEDs. Then, the in situ-generated α-carbonyl carbon radical was trapped by the alkenes, producing a benzylic radical intermediate 17, which was oxidize to the corresponding carboncation intermediate 18 by another SET, thus finishing the photocatalytic cycle. The intermediate 18 was then captured by a small amount of water to achieve the desired product. The authors also claim that the Cu(II) salt acted as an oxidant and nucleophile reagent carrier [34].
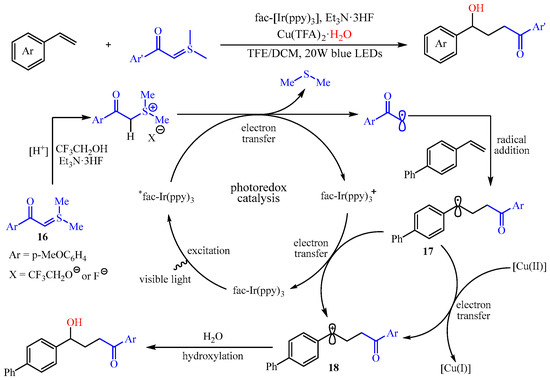
Scheme 20. Radical addition/hydroxylation reaction of alkenes, sulfur ylides, and water.
Huo and co-workers [35] found that the aromatic carboxylic acid oxime esters could be employed as bifunctional agents for the difunctionalization of aryl alkenes with (Ir[dF(CF3)ppy]2(dtbpy))PF6 as the catalyst (Scheme 21). Both O-centered radicals and N-centered radicals were generated in the meantime through the photosensitized Energy Transfer Catalysis (EnT) strategy-promoted O–N bond homolysis event [36]. With this strategy, a series of complex vicinal amino alcohols were facilely fabricated in one-step synthesis through the EnT-promoted 1,2-bifunctionalization of alkenes to decorate both C–N and C–O bonds synchronously. The substrate scope of this reaction proved to be broad, and an array of oxyaminated products were constructed in 40–79% yield. In addition, the synthetic utility of this transformation was further extended to the late-stage modification of complicated alkenes, such as the functionalization of several drug-related molecules and bioactive natural products.
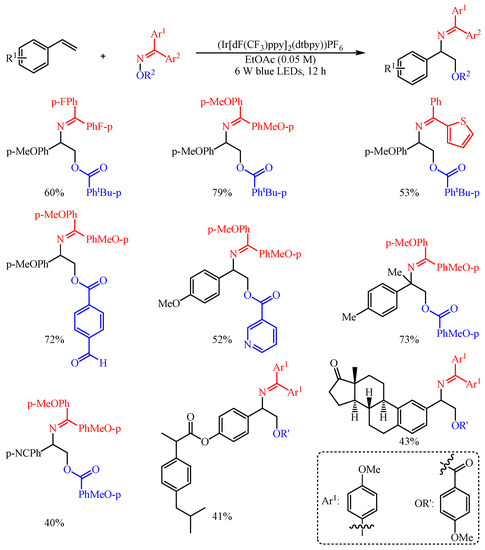
Scheme 21. Intermolecular oxyimination of alkenes with aromatic carboxylic-acid oxime esters.
References
- Klauck, F.J.R.; Yoon, H.; James, M.J.; Lautens, M.; Glorius, F. Visible-Light-Mediated Deaminative Three-Component Dicarbofunctionalization of Styrenes with Benzylic Radicals. ACS Catal. 2019, 9, 236–241.
- Cabrera-Afonso, M.J.; Sookezian, A.; Badir, S.O.; El Khatib, M.; Molander, G.A. Photoinduced 1,2-dicarbofunctionalization of alkenes with organotrifluoroborate nucleophiles via radical/polar crossover. Chem. Sci. 2021, 12, 9189–9195.
- Wang, X.F.; Guo, X.Y.; Liu, T. Two Co(II)-based Coordination Polymers Constructed from π-electron-rich Polycarboxylate Aryl Ether Ligand: Structural Insights and Photocatalytic Dye Degradation. Chin. J. Struct. Chem. 2021, 40, 722–728.
- Huang, H.L.; Xu, J.; Fan, Y.X.; Su, Q.Q.; Du, J.Y.; Zhang, R.F.; Wang, Y.L.; Hu, H.; Gao, F. Visible-Light-Induced Difunctionalization of Alkenyl Ketones with α-Carbonyl Alkyl Bromide: Concomitant Installation of C-C Bonds. J. Org. Chem. 2022, 87, 14093–14102.
- Mao, L.L.; Zhou, A.X.; Zhu, X.H.; Peng, H.; Quan, L.X.; Wan, J.P.; Yang, S.D. Visible-Light-Mediated Tandem Difluoromethylation/Cyclization of Alkenyl Aldehydes toward CF2H-Substituted Chroman-4-one Derivatives. J. Org. Chem. 2022, 87, 12414–12423.
- Lei, Z.R.; Wei, S.Q.; Zhou, L.J.; Zhang, Z.X.; Lopez, S.E.; Dolbier, W.R. Photocatalytic difluoromethylarylation of unactivated alkenes via a (hetero)aryl neophyl-like radical migration. Org. Biomol. Chem. 2022, 20, 5712–5715.
- Chen, H.; Ye, J.L.; Huang, P.Q. Chemoselective direct reductive trifluoromethylation of amides: A flexible access to functionalized alpha-trifluoromethylamines. Org. Chem. Front. 2018, 5, 943–947.
- Jang, J.; Kim, D.Y. Synthesis of Trifluoromethylated 4H-1-Benzopyran Derivatives via Photocatalytic Trifluoromethylation/Oxidation/Conjugate Addition, and Cyclization Sequences of Vinyl Phenols. Asian J. Org. Chem. 2022, 11, e202200052.
- Guan, Y.Q.; Min, X.T.; He, G.C.; Ji, D.W.; Guo, S.Y.; Hu, Y.C.; Chen, Q.A. The serendipitous effect of KF in Ritter reaction: Photo-induced amino-alkylation of alkenes. Iscience 2021, 24, 102969.
- Zhang, S.; Gutierrez-Bonet, A.; Molander, G.A. Merging Photoredox PCET with Ni-Catalyzed Cross-Coupling: Cascade Amidoarylation of Unactivated Olefins. Chem 2019, 5, 339–352.
- Feng, Z.; Xiao, Y.L.; Zhang, X.G. Palladium-catalyzed phosphonyldifluoromethylation of alkenes with bromodifluoromethylphosphonate. Org. Chem. Front. 2016, 3, 466–469.
- Yang, Q.; Li, C.; Qi, Z.C.; Qiang, X.Y.; Yang, S.D. Photocatalyzed Intermolecular Aminodifluoromethylphosphonation of Alkenes: Facile Synthesis of alpha,alpha-Difluoro-gamma- aminophosphonates. Chem. Eur. J. 2018, 24, 14363–14367.
- Li, D.K.; Mao, T.T.; Huang, J.B.; Zhu, Q. Copper-Catalyzed Bromodifluoroacetylation of Alkenes with Ethyl Bromodifluoroacetate. J. Org. Chem. 2018, 83, 10445–10452.
- Luo, X.W.; Zhang, B.; Xi, C.J. Photoredox-catalyzed hydroxydifluoroacetylation of alkenes with FSO2CF2CO2Me and H2O: Simple synthesis of CF2CO2Me-containing alcohols and difluorolactones. Green Chem. 2021, 23, 2324–2328.
- Zou, S.; Luo, X.W.; Chen, C.; Xi, C.J. Photoredox-catalyzed fluorodifluoroacetylation of alkenes with FSO2CF2CO2Me and Et3N center dot 3HF. Org. Biomol. Chem. 2022, 20, 3726–3730.
- Ge, H.; Wu, B.; Liu, Y.; Wang, H.; Shen, Q. Synergistic Lewis Acid and Photoredox-Catalyzed Trifluoromethylative Difunctionalization of Alkenes with Selenium Ylide-Based Trifluoromethylating Reagent. ACS Catal. 2020, 10, 12414–12424.
- Zhang, X.H.; Cao, Y.W.; Chen, Q.Y.; Shen, C.R.; He, L. Recent Progress in Homogeneous Reductive Carbonylation of Carbon Dioxide with Hydrogen. Acta Phys.-Chim. Sin. 2021, 37, 20070052.
- Wang, Q.; Nilsson, T.; Eriksson, L.; Szabo, K.J. Sulfenofunctionalization of Chiral alpha-Trifluoromethyl Allylboronic Acids: Asymmetric Synthesis of SCF3, SCF2R, SCN and SAr Compounds. Angew. Chem. Inter. Ed. 2022, 61, e202210509.
- Dagousset, G.; Simon, C.; Anselmi, E.; Tuccio, B.; Billard, T.; Magnier, E. Generation of the SCF3 Radical by Photoredox Catalysis: Intra- and Intermolecular Carbotrifluoromethylthiolation of Alkenes. Chem. Eur. J. 2017, 23, 4282–4286.
- Zhang, P.B.; Li, W.W.; Qu, W.L.; Shu, Z.G.; Tao, Y.Y.; Lin, J.M.; Gao, X. Copper and Photocatalytic Radical Relay Enabling Fluoroalkylphosphorothiolation of Alkenes: Modular Synthesis of Fluorine-Containing S-Alkyl Phosphorothioates and Phosphorodithioates. Org. Lett. 2021, 23, 9267–9272.
- Zhang, P.B.; Yu, G.; Li, W.W.; Shu, Z.G.; Wang, L.Y.; Li, Z.T.; Gao, X. Copper-Catalyzed Multicomponent Trifluoromethylphosphorothiolation of Alkenes: Access to CF3-Containing Alkyl Phosphorothioates. Org. Lett. 2021, 23, 5848–5852.
- Wang, H.M.; Bellotti, P.; Zhang, X.L.; Paulisch, T.O.; Glorius, F. A base-controlled switch of SO2 reincorporation in photocatalyzed radical difunctionalization of alkenes. Chem 2021, 7, 3412–3424.
- Zhao, X.J.; Chang, Y.D.; Chen, W.J.; Wu, Q.S.; Pan, X.Y.; Chen, K.F.; Weng, B. Recent Progress in Pd-Based Nanocatalysts for Selective Hydrogenation. ACS Omega 2022, 7, 17–31.
- Guyon, H.; Chachignon, H.; Cahard, D. CF3SO2X (X = Na, Cl) as reagents for trifluoromethylation, trifluoromethylsulfenyl-, -sulfinyl- and -sulfonylation. Part 1: Use of CF3SO2Na. Beilstein J. Org. Chem. 2017, 13, 2764–2799.
- Tang, K.; Chen, Y.X.; Guan, J.P.; Wang, Z.J.; Chen, K.; Xiang, H.Y.; Yang, H. Visible-light-promoted olefinic trifluoromethylation of enamides with CF3SO2Na. Org. Biomol. Chem. 2021, 19, 7475–7479.
- Tanaka, S.; Nakayama, Y.; Konishi, Y.; Koike, T.; Akita, M. Fluoroalkanesulfinate Salts as Dual Fluoroalkyl and SO2 Sources: Atom-Economical Fluoroalkyl-Sulfonylation of Alkenes and Alkynes by Photoredox Catalysis. Org. Lett. 2020, 22, 2801–2805.
- Liang, S.S.; Wei, J.J.; Jiang, L.Q.; Liu, J.; Mumtaz, Y.; Yi, W.B. Photocatalyzed Dual-Oxidative Trifluoromethylthio-Trifluoromethylation of Alkenes with CF3SO2Na. CCS Chem. 2021, 3, 265–273.
- Wang, L.; Cheng, P.; Wang, X.H.; Wang, W.; Zeng, J.G.; Liang, Y.; Reiser, O. Visible-light promoted sulfonamidation of enol acetates to alpha-amino ketones based on redox-neutral photocatalysis. Org. Chem. Front. 2019, 6, 3771–3775.
- Mo, J.N.; Yu, W.L.; Chen, J.Q.; Hu, X.Q.; Xu, P.F. Regiospecific Three-Component Aminofluorination of Olefins via Photoredox Catalysis. Org. Lett. 2018, 20, 4471–4474.
- Barata-Vallejo, S.; Lantano, B.; Postigo, A. Recent Advances in Trifluoromethylation Reactions with Electrophilic Trifluoromethylating Reagents. Chem. Eur. J. 2014, 20, 16806–16829.
- Yasu, Y.; Koike, T.; Akita, M. Three-component Oxytrifluoromethylation of Alkenes: Highly Efficient and Regioselective Difunctionalization of C=C Bonds Mediated by Photoredox Catalysts. Angew. Chem. Int. Ed. 2012, 51, 9567–9571.
- Zhou, F.; Cheng, Y.; Liu, X.P.; Chen, J.R.; Xiao, W.J. A visible light photoredox catalyzed carbon radical-mediated generation of ortho-quinone methides for 2,3-dihydrobenzofuran synthesis. Chem. Commun. 2019, 55, 3117–3120.
- Hirata, G.; Shimada, T.; Nishikata, T. Organo-photoredox-Catalyzed Atom-Transfer Radical Substitution of Alkenes with α-Carbonyl Alkyl Halides. Org. Lett. 2020, 22, 8952–8956.
- Yan, D.M.; Xu, S.H.; Qian, H.; Gao, P.P.; Bi, M.H.; Xiao, W.J.; Chen, J.R. Photoredox-Catalyzed and Copper(II) Salt-Assisted Radical Addition/Hydroxylation Reaction of Alkenes, Sulfur Ylides, and Water. ACS Catal. 2022, 12, 3279–3285.
- Li, J.; Yuan, Y.; Bao, X.; Sang, T.; Yang, J.; Huo, C. Visible-Light-Induced Intermolecular Oxyimination of Alkenes. Org. Lett. 2021, 23, 3712–3717.
- Hu, X.Q.; Chen, J.; Chen, J.R.; Yan, D.M.; Xiao, W.J. Organophotocatalytic Generation of N- and O-Centred Radicals Enables Aerobic Oxyamination and Dioxygenation of Alkenes. Chem. Eur. J. 2016, 22, 14141–14146.
More