Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Xuyao Zhang and Version 2 by Rita Xu.
Immunotherapies including adaptive immune checkpoint inhibitors (ICIs) and chimeric antigen receptor (CAR) T cells, have developed the treatment of cancer in clinic, and most of them focus on activating T cell immunity. Although these strategies have obtained unprecedented clinical responses, only limited subsets of cancer patients could receive long-term benefits, highlighting the demand for identifying novel targets for the new era of tumor immunotherapy. Innate immunity has been demonstrated to play a determinative role in the tumor microenvironment (TME) and influence the clinical outcomes of tumor patients.
- innate immunity
- cancer therapy
- immune checkpoint inhibitors
1. Introduction
Cancer is one of the main leading causes of disease-associated death and it often results from genotoxic and non-genotoxic carcinogens [1]. Surgery, chemotherapy, radiotherapy and tumor immunotherapy are the principle treatment for tumors. Surgery is the treatment of choice for most solid tumors, but cannot be used for hematologic tumors and metastases and subclinical metastases of solid tumors. Compared to surgical treatment, chemotherapy is a systemic treatment for primary, metastatic and subclinical metastases of solid tumors as well as hematologic tumors. However, the poor selectivity of chemotherapeutic agents limits its clinical application. Radiation therapy protects tissue that is not affected by the tumor, produces less damage and does not require hospitalization, but the application of radiotherapy depends on the type, size and location of tumors. Compared with these traditional therapies, tumor immunotherapy can kill tumor cells by activating the host’s immune system [2][3][2,3]. The discipline of oncology has been undergoing an unprecedented revolution due to the massive advancement of immunotherapy in the treatment of cancer [4]. Infiltrating into the TME, immune cells assist in tuning the development of tumors [5][6][5,6]. In adaptive immunity, T cells participate in immune responses that are cell-mediated immune responses [7][8][9][7,8,9]. The treatment of solid tumors has been revolutionized by ICIs targeting PD-1/PD-L1. Blocking the PD-1/PD-L1 axis could enhance T cell infiltration and revitalize exhausted cytotoxic T cells, presenting impressive therapeutic efficacy in various cancers [10]. Interestingly, PD-L1 expression and sensitivity to immunotherapy of tumor cells are subject to metabolic reprogramming. For example, mitochondrial oxidative phosphorylation (OXPHOS) can regulate PD-L1 levels. Tumor-selective OXPHOS suppression nanoparticles have been reported to reactivate immunotherapy and may open a new therapeutic window for patients [11]. Furthermore, CAR T cells have shown impressive efficacy in the treatment of hematologic malignancies [12][13][14][12,13,14]. As the mechanisms of tumor development and treatment are further explored, many new strategies have also emerged. For instance, oxidative stress and reactive oxygen species (ROS) contribute to the development of cancer growth [15]. Myrrh, a traditional remedy, could be a possible therapeutic agent for cancer because of the powerful antioxidant activity of its extracts [15]. Pyroptosis is a form of programmed cell death, which is associated with tumor genesis, and therapy response. Inflammasomes are multimolecular complexes containing pattern recognition receptors that can recruit the adaptor protein containing apoptosis-associated speck-like protein containing a caspase recruitment domain and activate caspase-1. To date, six members of the gasdermin (GSDM) family have been identified, namely GSDMA, GSDMB, GSDMC, GSDMD, GSDME (DFNA5) and DFNB59. Inflammasomes can activate caspase-1 that can cleave GSDMD to induce pyroptosis [16]. It is reported that the virus-like particle can trigger the formation of an AIM2 inflammasome that can induce GSDMD-mediated pyroptosis, thus enhancing antitumor immunity [17]. The primary goal of the currently approved immunotherapies is to active T cell immunity, but innate immunity also has significant anti-tumor potential. The desire to identify novel targets in innate immunity has emerged in the new era of immunotherapy.
In the innate immune system, myeloid-derived suppressor cells (MDSCs), macrophages, neutrophils, natural killer (NK) cells, dendritic cells (DCs), mast cells (MCs) and helper innate lymphoid cells (ILCs) have important regulatory effects on tumor progression [18]. Some innate immune cells have the capacity for detecting and eliminating tumor cells through various mechanisms, such as intrinsic cytotoxicity of NK cells and macrophage phagocytosis. The interaction of the innate and adaptive immunity is exemplified by the uptake of tumor antigens by antigen-presenting cells (APCs), which results in cross-presentation and the priming of CD8+ T lymphocytes [19]. Innate immune cells are also involved in effector responses through antibody-dependent cellular phagocytosis (ADCP) and antibody-dependent cell cytotoxicity (ADCC) [20].
2. Innate Immunity in TME
Composed of MDSCs, macrophages, neutrophils, NK cells, DCs, MCs and helper ILCs, the innate immunity plays a crucial role in defense against tumors (Figure 1). For example, macrophages can activate the tumor-killing activity of T cells through antigen cross-presentation. Antigen cross-presentation by macrophages can be achieved through the cytosolic and vacuolar pathways. In the cytosolic pathway, proteins are transported to the cell membrane, where they are degraded by the proteasome. Subsequently, the derived peptides are transferred via the transporter associated with antigen presentation to the endoplasmic reticulum (ER), where they are processed by aminopeptidases of the ER, or brought back to the antigen-containing endosome to be processed by insulin-regulated aminopeptidases. In this process, the peptide is loaded on the MHC-I. The other major cross-presentation pathway is the vacuolar pathway, in which proteins are processed by endosomal/lysosomal proteases and loaded on MHC-I within the endosomal/lysosomal compartments. The resulting peptide MHC-I molecular complex is recognized by CD8+ T cells expressing specific T cell receptors (TCRs). The signal from TCR recognition of antigen is transduced into CD8+ T cells, which triggers cytotoxic effects [21]. Neutrophils can promote tumor proliferation and invasion by secreting proteases, and can also inhibit tumor progression by secreting H2O2, based on the type, stage and location of the tumors [22]. NK cells achieve anti-tumor effects by mediating ADCC, and MCs can promote tumors or fight tumors by releasing cytokines, chemokines, eicosanoids, proteoglycans and biogenic amines [23][24][23,24]. A comprehensive understanding of innate immune cell function in the TME is vital for the design of effective cancer immunotherapy.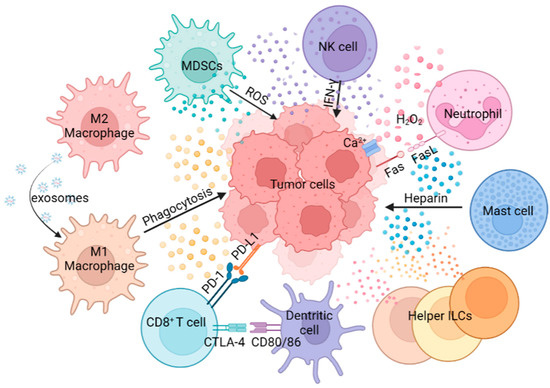
Figure 1. The role of innate immune cells in tumor progression. Innate immune cells show multiple roles in tumor progression through antigen presentation, phagocytosis, secretion of cytokines, direct killing effect, etc.