Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Emilio Mateev and Version 2 by Conner Chen.
With the significant growth of patients suffering from neurodegenerative diseases (NDs), novel classes of compounds targeting monoamine oxidase type B (MAO-B) are promptly emerging as distinguished structures for the treatment of the latter. As a promising function of computer-aided drug design (CADD), structure-based virtual screening (SBVS) is being heavily applied in processes of drug discovery and development. The utilization of molecular docking, as a helping tool for SBVS, is providing essential data about the poses and the occurring interactions between ligands and target molecules.
- molecular docking
- structure-based drug design
- MAO-B inhibitors
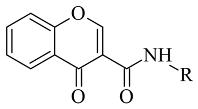
1. Chalcones
Chalcone-based compounds (Figure 13) have various therapeutic applications considering the diverse pharmacological targets they could affect [1][72]. Recently, the design and synthesis of MAO-B inhibitors containing a chalcone scaffold have rapidly been growing [2][73]. A major scientific effort in that area has been achieved by Bijo Mathew, confirmed by his involvement in numerous published papers concerning the MAO-B activity of chalcone derivatives. Various PDB structures have been applied in the reported docking protocols. Mathew et al. discussed the synthesis and molecular docking of eleven chlorinated thienyl chalcones (Table 12, item 1) containing MAO-B and MAO-A inhibitory effects [3][74].
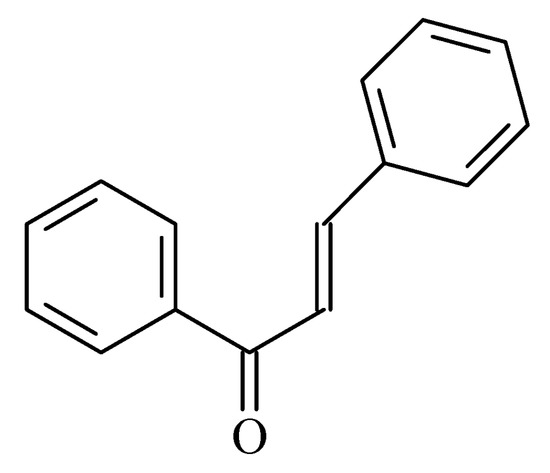
Figure 13.
Chemical structure of chalcone.
Table 12.
Structures of the reported chalcones evaluated as MAO-B inhibitors utilizing various docking software and PDB codes.
| S. No. | Structure | MAO-B Inhibitor Activity | Docking Program | PDB Code | Ref. |
|---|---|---|---|---|---|
| 1. |  |
Ki 5.52—0.49 (μM) | AutoDock 4.2 | 2BYB | [3][74] |
| 2. |  |
Ki 0.11—9.30 (μM) | AutoDock 4.2 | 2BYB | [4][75] |
| 3. | 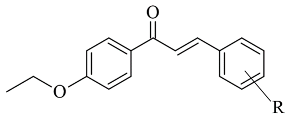 |
IC50 0.68—28.50 (μM) | Glide | 2V5Z | [5][76] |
| 4 | 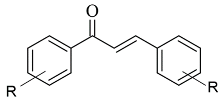 |
IC50 0.0029—13.2 (μM) | AutoDock Vina | 4A79 | [6][77] |
| 5. | 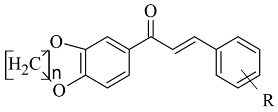 |
IC50 0.00021—0.068 (μM) | Glide | 2V5Z | [7][78] |
The authors used AutoDock4.2 as software for the molecular simulation and X-ray structure of hMAO-B in complex with deprenyl (PDB: 2BYB) obtained from the Protein Data Bank. Water molecules were deleted and the covalent bond between the co-crystallized ligand and N5-atom of FAD in the structure of 2BYB was corrected. The center of the grid was set at N5 atom of FAD. The thiophene moiety was placed in the substrate cavity, while the aromatic ring of the chalcones interacted with Tyr435 through Van der Waals bonds (Figure 24).
Further stabilization via π-π interactions between the ligands and the aromatic cage was noted. It was concluded that the ethyl substitution in the phenyl ring led to higher MAO-B affinity. The same group published a paper of the synthesis and computational studies of brominated thienyl chalcones (Table 12, item 2) [4][75]. The utilized docking protocol of this study corresponds to the one described in the previous case. Among ethyl, methyl, methoxy, and dimethylamine groups placed at the para-position of the brominated thienyl chalcones, the dimethylamino moiety showed the best potency and selectivity. Furthermore, the docking software predicted that the indicated groups were projected into the entrance cavity. Further examination showed the dimethylamino moiety, as the bulkiest group, spanned both the entrance and the substrate pockets. π-π interactions between the aromatic ring of the chalcone and the “aromatic cage” were also considered. Glide docking, for the evaluation of the MAO inhibitory activity of eleven ethoxy substituted chalcones (Table 12, item 3), was applied by Lakshminarayanan et al. [5][76]. The ligands were first synthesized, and the virtual screening was carried out on an X-ray MAO-B structure (PDB: 2V5Z). Hydrophobic interactions between the ligands and the amino acids Ile199, Ile316, and leu171 were examined (Figure 35).

Figure 35. The active amino residues of MAO-B are presented as an cyan blue, and the ethoxylated chalcone is depicted in red [5].
The active amino residues of MAO-B are presented as an cyan blue, and the ethoxylated chalcone is depicted in red [76].
Compound 1-(4-ethoxyphenyl)-3-(4-fluorophenyl)prop-2-en-1-one displayed the best MAO-B inhibitory potential. The acquired docking poses show that the methyl group of the p-ethoxy moiety was placed in the aromatic cage of the MAO-B structure. When subjected to virtual screening in the active site of MAO-A, the interaction with FAD was absent due to a steric hindrance.
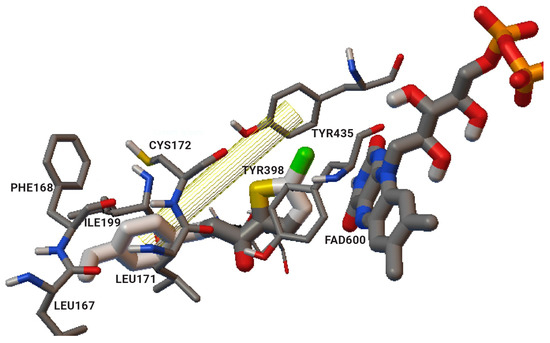
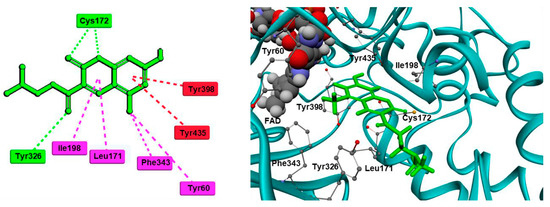
John Oh et al. [6][77] studied the MAO-B inhibition effect of various natural and synthetic chalcones (Table 12, item 4). The molecular docking simulation was carried out by AutoDock Vina using the 3D structure of MAO-B with co-crystallized pioglitazone (PDB: 4A79). Further docking studies towards MAO-A and AChE receptors were performed, and it was discussed that the analyzed chalcones have higher selectivity towards MAO-B. The dimethoxy chalcone oxime with fluoro substitution in the acetophenone moiety showed the most prominent selectivity and blocking activity towards MAO-B. It was observed that the high MAO-B activity was due to the hydrophobic π-π interactions between Tyr326 and the ligand. Furthermore, the formation of a hydrogen bond between the carbonyl oxygen of chalcones and Cys172 of MAO-B increased the binding affinity of the ligand (Figure 46).
The design, synthesis, and biological evaluation of oxygenated chalcones (Table 12, item 5) were reported by Parambi et al. [7][78]. The crystal structure of MAO-B was downloaded from PDB: 2V5Z. The Schrödinger suite was used for protein preparation, and eight molecules of water were preserved in the active site of MAO-B. The QM polarized ligand docking from Schrödinger Suite was applied as docking software. The docking study of the most active and selective oxygenated chalcone demonstrated that the benzoxazole group was close to the FAD and moderate–strong π-π stacking interactions with Ile199, Tyr398, and Tyr435 formed (Figure 57). An additional hydrogen bond was established between Tyr326 and the hydrogen bond acceptor of the ligand, which further confirms the important role of Tyr326 for the MAO selectivity.
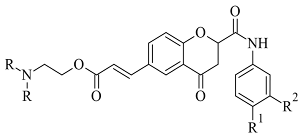
2. Coumarins
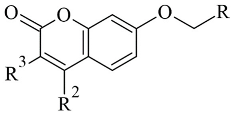
Coumarins are structural isomers of chromones and were investigated for MAO, cholinesterase, and aromatase antagonizing activities (Figure 68). The latter multitarget properties were utilized for the treatment of neurodegenerative disorders [8][79]. Recent papers have demonstrated the selectivity and potency of coumarins as MAO-B inhibitors [9][80].
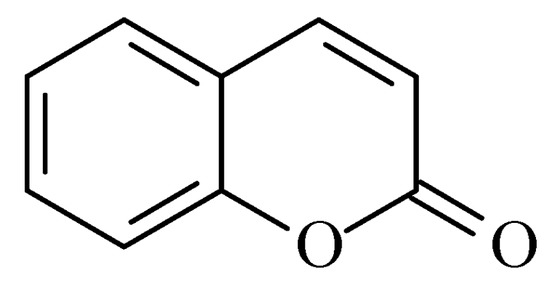
Figure 68.
Chemical structure of coumarin.
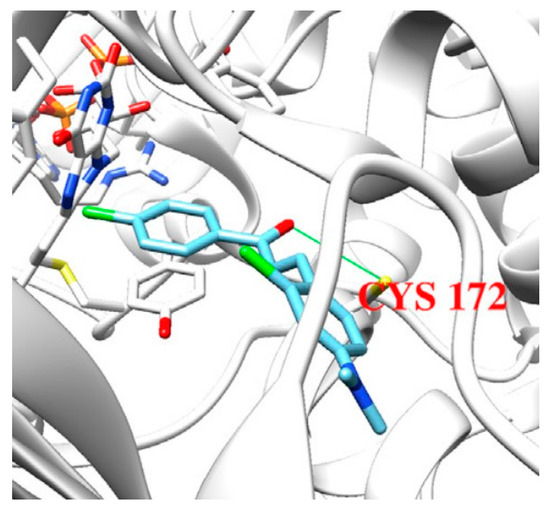
Joubert et al. reported the synthesis and virtual screening results of 7-substituted coumarin derivatives (Table 23, item 1) [10][81]. Molecular operating environment (MOE) software was applied for the docking study, and hMAO-B crystal structure with co-crystallized 7-(3-chlorobenzyloxy)-4-(methylamino)methylcoumarin (PDB: 2V61) was used. The obtained data show that the top solutions situated their coumarin moiety in the polar region of the substrate cavity (Figure 79).

Moreover, a hydrogen bond between the carbonyl group of most compounds and Cys172 was observed. Van der Waals interactions between benzyl and the N-benzylpiperidine side chain of the molecules and the hydrophobic entrance cavity further stabilized the complex.
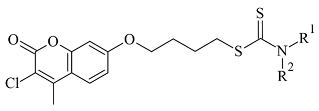
Another study reported the synthesis and determination of the MAO-B inhibitory activity of coumarin–dithiocarbamate hybrid compounds [11][82]. The research group applied Molecular Operating Environment (MOE, version 2008.10) software for the molecular modeling. The study concluded that the coumarin moiety (Table 23, item 2) was in the substrate cavity and further stabilized by binding to Tyr60, Phe343, Tyr398, and Tyr435. The dithiocarbamate part of the molecule was situated in the entrance cavity. The most active compound of the coumarin–dithiocarbamate explored class was with an IC50 value of 0.101 µM. Eight compounds isolated from glycyrrhiza uralens, also known as Chinese licorice, were subjected to a molecular study and examined for their monoamine oxidase and cholinesterase inhibitory activities [12][83]. It was found that liquiritigenin has a moderate MAO-B inhibitory constant (IC50 = 0.098 µM). The virtual screening was carried out with AutoDock Vina, and MAO-B with co-crystallized pioglitazone (PDB: 4A79) was downloaded from PDB. The active site grid was set as defined by the binding site of co-crystallized pioglitazone. The work demonstrated that in the MAO-B active site, no hydrogen bonds were formed. However, a promising correlation between in vitro and in-silico data was obtained.
Repsold et al. analyzed the MAO-B and AChE inhibitory properties of new lead compounds with coumarin moiety (Table 23, item 3) [13][84]. Piperidine, morpholine, thiophene, and erucic acid were used for the conjugation with coumarin. The software utilized in that research was Accelrys® Discovery Studio® V3.1.1, and an X-ray of MAO-B with the co-crystallized selective inhibitor (7-(3-chlorobenzyloxy)-4-(methylamino) methyl coumarin) was taken from PDB. The virtual screening indicated a reasonable MAO-B blocking activity of the coumarin–morpholine ether. It demonstrated a crucial p-interaction with Tyr398 and hydrogen bond with Cys172.
Discovery Studio 2016 was utilized in the molecular docking of novel Mannich bases of 3-acetyl-7-hydroxyl coumarins (Table 23, item 4) by Tao et al. [14][86]. After the initial in vitro determination of MAO-B activity, two compounds showed promising antagonism towards the enzyme and were further evaluated. The coumarin moiety was orientated in the substrate cavity between Tyr398 and Tyr435. Moreover, the complex was further supported by a hydrogen bond between Cys172 and the oxygen atom of the lactone ring (Figure 810).
Table 23.
Structures of the reported coumarins evaluated as MAO-B inhibitors utilizing various docking software and PDB codes.

Rauhamaki et al. designed a virtual library of 3-phenylcoumarin derivatives (Table 23, item 5) and subjected them to a virtual screening using Glide [15][87]. The most promising compounds were synthesized and assessed in vivo for MAO-B activity. From the computational study, the importance of the 3-phenyl moiety of the coumarins for the MAO-B inhibitory activity was analyzed. The hydrophobic interaction of the coumarin and 3-phenyl ring with the substrate pocket was established as an essential part of the MAO-B inhibitory activity (Figure 911).
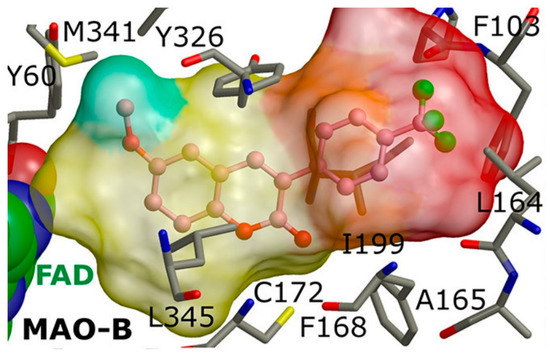
Interestingly, the 3-phenylcoumarins were reversed in the active site compared to the validated co-crystallized ligands. Moreover, the authors discussed an atypical hydrogen bond of the 2C-carbonyl group with the thiol moiety of Cys172; however, the latter interaction did not inflict any alterations.
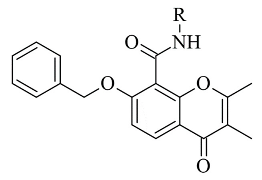
3. Chromones
Recently published works have demonstrated the prominent chromone scaffold (Figure 102) for the design of novel MAO-B inhibitors [16][88]. The majority of the chromone derivatives demonstrated high selectivity towards MAO-B, which has been explained with the smaller active gauge in MAO-A [17][89]. An emphasis was placed on the chromone-3-phenylcarboxamide moiety [18][90].
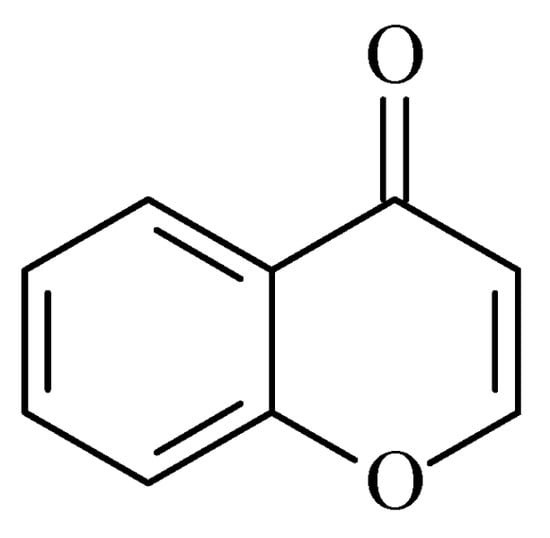
Figure 102.
Chemical structure of chromone.
Reis et al. reported the synthesis and the molecular docking of a small library of chromone-3-phenylcarboxamides (Table 34, item 1) with MAO-B blocking activity [19][91]. Glide was utilized as docking software for the virtual screening of the potent chalcones. Interactions of the chalcone derivatives with Phe102 and Tyr435 have been noted (Figure 113).
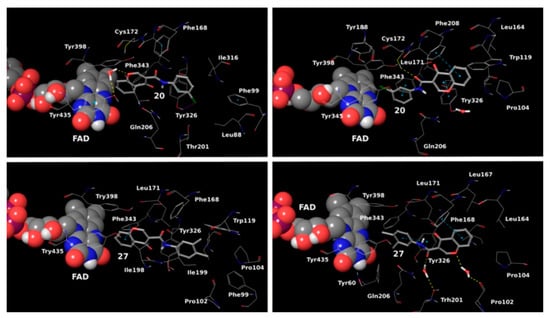
Table 34. Structures of the reported chromone derivatives evaluated as MAO-B inhibitors utilizing various docking software and PDB codes.

Additionally, the water-bridged bond of the carbonyl oxygen with Tyr201 was observed. The paper examined the steric hindrance when the bulky structure was set into the active site of MAO-B. The latter ligands lacked strong interactions and, therefore, were assessed as weaker inhibitors. The work further discussed the solvent effect after MD simulations and trajectories inspections. Moderate correlation with the experimental data was observed. Another paper published by Reis et al. discussed the synthesis and the screening of selective MAO-B chromone derivatives (Table 34, item 2) [20][92]. The most promising compounds were virtually evaluated using the Glide module in Schrödinger Suite 2016-3. The grid was centered at N5 of FAD. As presumed, the phenylcarboxamide moiety was in the aromatic cage. The larger ligand pocket in the active site of MAO-B is accountable for facilitated ligand fitting. MAO-A has a smaller active cavity which leads to a steric hindrance and therefore inability in the formation of stable interactions with the ligands.
Genetic Optimization of Ligand Docking (GOLD) 3.0.1 was applied for the virtual screening of novel 7-benzyloxy-8-carboxamide-chromone derivatives (Table 34, item 3) with selective MAO-B inhibitory activity [21][93]. The authors noted that the occurrence of numeral hydrogen bonds between the functional groups of the chromone derivatives and the amino residues from the active site drastically increased the potency of the ligands. The hydrogen bonds were explained with the presence of several groups (mainly oxygen and nitrogen atoms) propagating these types of interactions. The molecular docking revealed the optimal amide aryl and benzyl orientations, in the active site, for a prominent MAO-B activity.
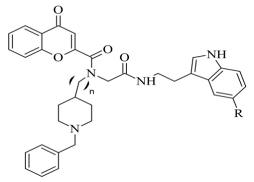
Virtual screening of newly synthesized chromone-donepezil hybrid compounds (Table 34, item 4) as carried out by Wang et al. [22][94]. The research group applied MOE 2008.10 as a docking program for the evaluation of the binding modes. The results displayed π-π interactions between the benzyloxy group of the ligands and the aromatic cage strictly with Tyr398 and Tyr435. An additional hydrogen bond formed between Tyr326 and the amide carbonyl of one of the compounds. Moreover, the quaternary nitrogen of the piperidine ring was involved in hydrogen bond interaction with Glu84.
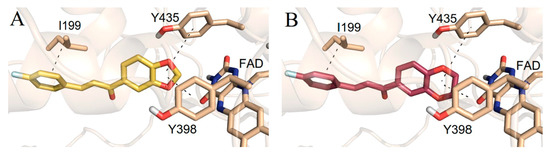
A docking study of chromone derivatives (Table 34, item 5) was carried out using Discovery Studio 3.1 [23][95]. X-ray structure of MAO-B was downloaded from a protein data bank (PDB: 2Z5X). It is noted that the most prominent molecules were 3-aminomethylidene-2,4-chromandiones. Two isomers of the most potent chromone with MAO-B blocking activity have been analyzed. As expected, π-π interaction of the chromone with Tyr398 and Tyr435 was observed (Figure 124).

Figure 124.
Active conformations of 3-aminomethylidene-2,4-chromandiones into the active site of
Furthermore, a notable hydrogen bond between the carbonyl group located at C2 of the chromone ring and Ile199 was examined. The molecular docking concluded that both cis and trans isomers display MAO-B inhibitory activity. Donepezil + chromone + melatonin hybrids (Table 34, item 6) were synthesized and virtually evaluated for their implementation in Alzheimer’s disease [24][96]. X-ray MAO-B with co-crystallized safinamide was used as a model target (PDB: 2V5Z). AutoDockVina was applied as docking software. π-π interactions were observed between the phenyl ring of the hybrid derivatives and Tyr60, Gly205, Phe343, and Tyr398 (Figure 135).
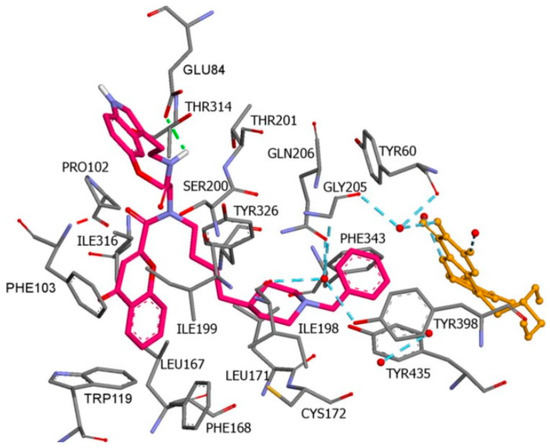
A hydrogen bond formed between the amide group and Glu84. The indole ring was situated at the beginning of the hydrophobic entrance pocket. The chromone moiety was positioned in the entrance cavity as well.