Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Drew Frase and Version 2 by Fanny Huang.
B-lymphocytes—typically appreciated for their canonical role in adaptive, humoral immunity—have emerged as critical regulators of bone remodeling. B-lymphocytes communicate with osteoclasts and osteoblasts through various cytokines, including IL-7, RANK, and OPG. In inflammatory conditions, B-lymphocytes promote osteoclast activation and differentiation. However, B-lymphocytes also possess immunomodulatory properties, with regulatory B-lymphocytes (Bregs) secreting TGF-β1 to restrain pathogenic osteoclastogenesis.
- osteoporosis
- B-lymphocytes
- inflammation
- bone remodeling
1. Introduction
Osteoporosis, a disorder characterized by reduced bone mass and the structural deterioration of osteogenic tissue, affects a significant proportion of people in the United States over 50 years old [1]. Considering a total of almost 10 million cases and an estimated 30 million at risk, osteoporosis is an ever-growing health concern [1]. In a meta-analysis conducted by Salari et al., using 86 studies across five countries, the global osteoporosis prevalence was 18.3%, with high rates observed in European and African countries [2]. In the United States, the age-adjusted prevalence of adults 50 years and older was almost 13% from 2017 to 2018, with an increased prevalence in men compared to women (19.6% and 4.4%, respectively) [3].
Osteoporosis most commonly manifests in the form of femoral neck or vertebral fractures, which can significantly reduce quality of life [4]. Fracture consequences include disability, extensive medical costs, and even increased mortality—underscoring the importance of understanding the mechanisms of osteoporosis and potential therapeutics [5]. In addition to direct physical consequences, the fiscal burden of osteoporosis and its concomitant fractures—both directly through therapeutic expenses and indirectly through missing school or work—impose substantial challenges to United States healthcare systems [6][7]. Economic forecasts suggest that by 2025, yearly fractures from osteoporosis will eclipse 3 million cases at a predicted cost of 25.3 billion dollars [8][9].
Recent research has begun to highlight the immune system’s critical role in the development and progression of osteoporosis, focusing mainly on soluble mediators, growth factors, and chemokines [10]. Emerging work has implicated both adaptive and innate immune system components, paying particular attention to their contribution to proinflammatory milieu [11][12][13]. In reference to these findings, Srivasta et al. proposed a novel term, “immunoporosis,” to stress the importance of immune cells in osteoporosis pathogenesis [14][15].
2. B-Lymphocytes
2.1. B-Lymphocytes and Bone Homeostasis in Osteoporosis
B-lymphocytes canonically represent the most significant component of the humoral adaptive immune system. After developing from hematopoietic stem cell (HSC) precursors in the bone marrow, mature B-lymphocytes’ primary role is to secrete antibodies that neutralize pathogens and potentiate the effector functions of other immune cells. B-lymphocytes play a direct role in antigen-dependent T-cell activation in the lymph node. Beyond their traditional roles in adaptive immunity, however, B-lymphocytes are increasingly being considered for their potential roles in diseases of bone remodeling. Given the anatomical proximity of the bone architecture to immune cell genesis in the bone marrow, osteoblasts, osteoclasts, and immune cells have long been thought to communicate via shared signaling mechanisms. Recent terms such as “osteoimmunology” and “immunoporosis” have explored the relationship between bone homeostasis and immune cells in osteoporosis-related diseases. However, little attention has been paid to the pathogenic disruptions to bone homeostasis driven specifically by B-lymphocytes [14][15].
In the bone marrow, B-lymphocyte development is highly dependent upon secreted factors derived from stromal cells and osteoblasts. During B-lymphocyte maturation, successful V(D)J recombination generates an immunoglobulin heavy chain (Ig-H-chain) as part of the pre-B-cell receptor (pre-BCR) [16]. Bruton’s tyrosine kinase (BTK) signaling appears to attenuate signals through the pre-BCR to limit proliferation [16]. Contrastingly, signaling through mature B-lymphocyte receptors (BCRs) leads to the activation of the phosphorylation of tyrosine residues on BTK [17]. Surprisingly, BTK signaling also plays a critical role in osteoclast differentiation [18][19]. Mice deficient in BTK signaling exhibited an osteopetrosis-like phenotype due to impaired bone resorption from deficient RANKL:BTK signaling in osteoclasts [18]. Additionally, when X-linked immunodeficient mice—with dysfunctional BTK signaling—were treated with receptor activator of nuclear factor kB (NF-Kb), osteoclast precursors failed to fuse into active multinucleated osteoclasts [19].
Over the last few decades, various studies have linked key cytokines and molecules to B-lymphocyte development and bone remodeling, including IL-7, receptor activator of nuclear factor kB (RANK), and osteoprotegerin [OPG]. Initial IL-7/IL-7R knockout studies observed that IL-7 transgenic mice had increased B-lymphocyte precursor levels, leading to a purported link between defects in B-lymphocyte development and bone mass [20]. Similar mouse studies knocking out RANK—the receptor for RANKL—led to fewer B-lymphocytes in lymph nodes [21]. RANK, RANKL, and OPG play a pivotal role in the differentiation and activation of osteoclasts. Shortly after IL-7 and RANK experiments began associating B-lymphocytes with bone remodeling, B-lymphocytes were shown to secrete OPG (Figure 1) [22]. OPG is a tumor necrosis factor (TNF) receptor family member; it is a molecular decoy receptor that binds RANKL, inhibiting RANKL:RANK-mediated osteoclastogenesis and preventing excessive bone resorption. Surprisingly, B-lymphocytes have been shown to produce roughly half of the total OPG produced in the bone marrow [23]. In mice with B-lymphocytes knocked out, their bone marrow was deficient in OPG, and osteoporotic bone was more prevalent than in controls [24]. Significantly, B-lymphocyte transplantation rescued the osteoporotic phenotype and improved bone marrow OPG levels [24]. In addition to osteoclastogenic suppression, B-lymphocytes also inhibit osteoblast differentiation via the secretion of C-C motif chemokine ligand 3 (CCL3) and TNF, which target extracellular signal-regulated kinase (ERK) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) to impair osteoblast differentiation [25].
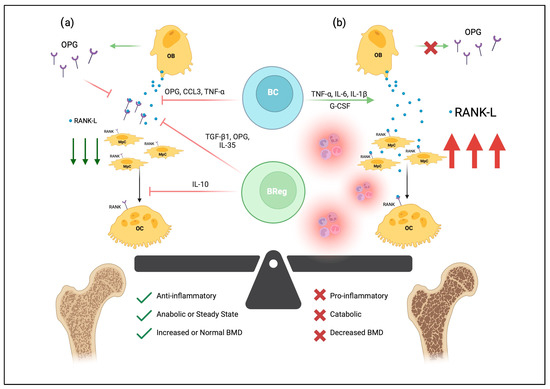
Figure 1. The effects of B-lymphocytes on bone homeostasis. B-lymphocytes (BC) and osteogenic precursors develop together in the bone marrow niche. In (a), osteoblasts (OB) can secrete RANKL, which binds RANK on the surfaces of monocyte-like progenitor cells (MpC) and stimulates differentiation and fusion into multinucleated osteoclasts (OC). RANKL also binds RANK on osteoclasts to promote survival and proliferation. In healthy bone, pro-osteoclastogenic RANKL is regulated by the osteoprotegerin (OPG) decoy receptor, as well as various soluble mediators secreted by OBs, BCs, and B-regulatory lymphocytes (Bregs). Thus, normal B-lymphocyte and osteogenic signaling generates an anti-inflammatory environment that maintains bone mineral density (BMD). In osteoporotic tissue, seen in (b), proinflammatory mediators drive osteoclastogenesis through increased expression of RANK-L, shifting the balance of bone metabolism toward catabolism and ultimately bone loss. Created with BioRender.com, accessed on 3 June 2023.
The role of B-lymphocytes in bone remodeling is context-dependent. B-lymphocytes secrete many factors critical in maintaining the bone architecture and share many cytokines with osteoclasts and osteoblasts. While B-lymphocytes play a critical role in suppressing osteoclasts via the secretion of OPG receptor decoys, inflammatory environments can redirect B-lymphocyte effects on bone remodeling toward bone resorption (Figure 1) [26]. In particular, IL-6, TNF-α, and IL-1β, are critical in driving the inflammatory pathophysiology of bone remodeling diseases [27]. In inflammatory conditions, B-lymphocytes have been shown to secrete RANKL, which stimulates the activation of osteoclasts [28][29][30]. Comparatively, RANKL knockout mice experienced greater protection against bone loss following ovariectomy than controls, while knockout in T-cells did not protect against bone loss after ovariectomy [30].
In addition to RANKL-mediated osteoclast activation, B-lymphocytes in inflammatory environments also secrete granulocyte colony-stimulating factor (G-CSF), which leads to osteoclast progenitor proliferation [31]. The production of both G-CSF and RANKL drives the proliferation and differentiation of osteoclasts, ultimately leading to bone resorption and loss of bone mass. G-CSF also appears to play a role in regulating neutrophil infiltration, which can lead to enhanced inflammation [31]. B-lymphocytes have been known to secrete IL-18 for years; however, the effect of IL-18 on bone remodeling was first reported to be anti-osteoclastogenic [32]. B-lymphocyte secretion of IL-18 was initially shown to upregulate OPG expression on osteoblastic cells, inhibiting osteoclastogenesis through the OPG/RANKL axis [32]. In a more recent study, however, B-lymphocyte secretion of IL-18 increased the surface expression of RANKL on T-lymphocytes, ultimately contributing to osteoclastogenesis [33]. In the latter study of osteoporotic women, IL-18 was increased compared to controls.
The contrasting roles of B-lymphocytes in osteoclastogenesis support the crucial role of B-lymphocytes in the balance of metabolic bone remodeling. Thus, disruptions of B-lymphocyte-mediated signaling via inflammatory environments are thought to drive catabolic bone resorption by promoting osteoclast differentiation and activation. Osteoporosis, in particular, appears to be driven in part by disrupted B-lymphocyte and bone homeostasis. In one postmenopausal osteoporosis study, women with osteoporosis had significantly fewer CD19+ B-lymphocytes than healthy controls [26]. Healthy controls also secreted less macrophage colony-stimulating factor (M-CSF) and had increased bone mineral density (BMD) in their lumbar spine [26]. Despite the association, it remains unclear whether reduced BMD impairs B-lymphocyte development or if impaired B-lymphocyte development reduces BMD.
2.2. Regulatory B-Lymphocytes (Bregs) in Osteoporosis
In addition to traditional antibody-secreting B-lymphocytes, B-regulatory lymphocytes (Bregs) are being increasingly recognized for their immunomodulatory role in bone homeostasis. Various Breg cytokines, including TGF-β1, IL-10, and IL-35 have been shown to modulate osteogenic differentiation. IL-35 is a ligand for the IL-35 receptor, which drives Breg differentiation via STAT1/STAT3 signaling pathways [34]. IL-35:IL-35-R binding also appears to suppress osteoclastogenesis by OPG secretion and subsequent RANKL downregulation [35].
Breg regulatory functions are largely attributed to the anti-inflammatory cytokine IL-10 [36][37]. Of note, CD19+ CD1dhi CD5+ [B10] Bregs were recently shown to secrete IL-10 and protect against osteoclastogenic pathogenesis [38]. In a recent animal model, IL-10 induced osteoblast differentiation by downregulating miR-7015-5p [39]. Besides driving osteoblast differentiation, IL-10 also suppresses osteoclast development. IL-10 directly impairs Ca2+ mobilization and NFATc1 signaling in osteogenic precursors, preventing the development of osteoclasts [40]. Furthermore, the adoptive transfer of a subset of Breg (B10) cells appeared to delay the onset of osteoporosis in an ovariectomized mouse model [41].
TGF-β1 is also a well-recognized anti-inflammatory cytokine and is also secreted by Bregs [42][43]. TGF-β1 and bone morphogenic proteins (BMP) constitute a critical developmental axis that signals traditionally via SMADS or nontraditionally via p38 MAPK pathways [42][43]. While the roles of TGF-β1 as a developmental morphogen are receiving increasing attention, fewer studies document the anti-inflammatory benefits of TGF-β1 in osteoporosis. The SMAD and MAPK pathways converge on signaling cascades that upregulate pro-osteoblastic factors such as Runt-related transcription factor 2 (RUNX2) [42]. As with IL-10, TGF-β1 also appears to decrease NFAT signaling and decreases RANK expression on osteoblasts [42]. TGF-β1 also decreases the expression of crucial osteoclastic genes cathepsin K and acid phosphatase 5, tartrate-resistant [42]. Thus, in osteoporotic conditions where TGF-β1 is reduced, RANKL expression would remain on osteoblasts, and RUNX2-directed osteoblastic differentiation would be reduced, both of which shift bone remodeling toward resorption.
Bregs represent an exciting new frontier in the field of osteoimmunology and immunoporosis. Currently, the anti-inflammatory mechanisms of Bregs, mediated via IL-10 and TGF-β1, offer promise in tempering proinflammatory environments that increase osteoclastogenesis and promote the pathogenic bone loss characteristic of osteoporosis. Current studies showing the positive protective effects of Bregs in bone remodeling are limited to animal models but have nonetheless been promising. Particularly in osteoporosis, it is imperative to shift the metabolic balance away from resorption.
References
- Office of the Surgeon General. Reports of the Surgeon General. Bone Health and Osteoporosis: A Report of the Surgeon General; Office of the Surgeon General (US): Rockville, MD, USA, 2004.
- Salari, N.; Ghasemi, H.; Mohammadi, L.; Hasan Behzadi, M.; Rabieenia, E.; Shohaimi, S.; Mohamadi, M. The global prevalence of osteoporosis in the world: A comprehensive systematic review and meta-analysis. J. Orthop. Surg. Res. 2021, 16, 609.
- Sarafrazi, N.; Wambogo, E.; Shepherd, J. Osteoporosis or Low Bone Mass in Older Adults: United States, 2017–2018; NCHS Data Brief No. 405 March 2021; National Center for Health Statistics (U.S.): Hyattsville, MD, USA, 2021.
- Lane, J.M.; Khan, R. Osteoporosis. Clin. Orthop. Relat. Res. 2000, 372, 139–150.
- Kemmak, A.R.; Rezapour, A.; Jahangiri, R.; Nikjoo, S.; Farabi, H.; Soleimanpour, S. Economic burden of osteoporosis in the world: A systematic review. Med. J. Islam. Repub. Iran 2020, 34, 154.
- Haussler, B.; Gothe, H.; Gol, D.; Glaeske, G.; Pientka, L.; Felsenberg, D. Epidemiology, treatment and costs of osteoporosis in Germany—The Bone EVA Study. Osteoporos. Int. 2007, 18, 77–84.
- Heydarpour, F.; Sari, A.A.; Mohebali, M.; Shirzadi, M.; Bokaie, S. Economic Burden of Cutaneous and Visceral Lishmaniasis in Iran in 2013. Iran J. Epidemiol. 2017, 12, 1–13.
- Burge, R.; Dawson-Hughes, B.; Solomon, D.; Wong, J.B.; King, A.; Tosteson, A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J. Bone Miner. Res. 2007, 22, 465–475.
- Roche, J.W.; Wenn, R.T.; Sahota, O.; Moran, C.G. Effect of comorbidities and postoperative complications on mortality after hip fracture in elderly people: Prospective observational cohort study. BMJ 2005, 331, 1374.
- Brunetti, G.; D’Amelio, P.; Mori, G.; Faienza, M.F. Editorial: Updates on osteoimmunology: What’s new on the crosstalk between bone and immune cells. Front. Endocrinol. 2020, 11, 74.
- Mundy, G.R. Osteoporosis and inflammation. Nutr. Rev. 2007, 65, S147–S151.
- Zupan, J.; Jers, M.; Marc, J. Osteoimmunology and the influence of proinflammatory cytokines on osteoclasts. Biochem. Med. 2013, 23, 43–63.
- Zhang, W.; Gao, R.; Rong, X.; Zhu, S.; Cui, Y.; Liu, H.; Li, M. Immunoporosis: Role of immune system in the pathophysiology of different types of osteoporosis. Front. Endocrinol. 2022, 13, 965258.
- Srivastava, R.K.; Dar, H.Y.; Mishra, P.K. Immunoporosis: Immunology of osteoporosis-role of T cells. Front. Immunol. 2018, 9, 657.
- Srivastava, R.K.; Sapra, L. The rising era of “Immunoporosis”: Role of immune system in the pathophysiology of osteoporosis. J. Inflam. Res. 2022, 15, 1667–1698.
- Hendriks, R.; Middendorp, S. The pre-BCR checkpoint as a cell-autonomous proliferation switch. Trends Immunol. 2004, 5, 249–256.
- Aoki, Y.; Isselbacher, K.J.; Pillai, S. Bruton tyrosine kinase is tyrosine phosphorylated and activated in pre-B lymphocytes and receptor ligated B cells. Proc. Natl. Acad. Sci. USA 1994, 91, 10606–10609.
- Shinohara, M.; Koga, T.; Okamoto, K.; Sakaguchi, S.; Arai, K.; Yasuda, H.; Takai, T.; Kodama, T.; Morio, T.; Geha, R.S.; et al. Tyrosine kinases BTK and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell 2008, 132, 794–806.
- Lee, S.H.; Kim, T.; Jeong, D.; Kim, N.; Choi, Y. The tec family tyrosine kinase BTK regulates RANKL-induced osteoclast maturation. J. Biol. Chem. 2008, 283, 11526–11534.
- Valenzona, H.O.; Pointer, R.; Ceredig, R.; Osmond, D.G. Prelymphomatous B-lymphocyte hyperplasia in the bone marrow of interleukin-7 transgenic mice: Precursor B cell dynamics, microenvironmental organization and osteolysis. Exp. Hematol. 1996, 24, 1521–1529.
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424.
- Yun, T.J.; Chaudhary, P.M.; Shu, G.L.; Frazer, J.K.; Ewings, M.K.; Schwartz, S.M.; Pascual, V.; Hood, L.E.; Clark, E.A. OPG/FDCR-1 a TNF receptor family member is expressed in lymphoid cells and is up-regulated by ligating CD40. J. Immunol. 1998, 161, 6113–6121.
- Walsh, M.C.; Choi, Y. Biology of the RANKL-RANK-OPG system in immunity, bone, and beyond. Front. Immunol. 2014, 5, 511.
- Li, Y.; Toraldo, G.; Li, A.; Yang, X.; Zhang, H.; Qian, W.P.; Weitzman, M.N. B-lymphocytes and T-cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood 2007, 109, 3839–3848.
- Sun, W.; Meednu, N.; Rosenberg, A.; Moreno-Rangel, J.; Wang, V.; Glanzman, J.; Owen, T.; Zhou, X.; Zhang, H.; Boyce, B.F.; et al. B-lymphocytes inhibit bone formation in rheumatoid arthritis by suppressing osteoblast differentiation. Nat. Commun. 2018, 9, 5127.
- Breuil, V.; Ticchioni, M.; Test, J.; Roux, C.H.; Ferrari, P.; Breittmayer, J.P.; Albert-Sabonnadiere, C.; Durant, J.; Perrenti, F.D.; Bernard, A.; et al. Immune changes in post-menopausal osteoporosis: The Immunos Study. Osteoporos. Int. 2010, 21, 805–814.
- Dar, H.Y.; Azam, Z.; Anupam, R.; Mondal, R.K.; Srivastava, R.K. Osteoimmunology: The nexus between bone and immune system. Front. Biosci. 2018, 23, 4600.
- Weitzmann, M.N. The Role of Inflammatory Cytokines, the RANKL/OPG axis, and the immunoskeletal interface in physiological bone turnover and osteoporosis. Scientifica 2013, 2013, 125705.
- Li, Y.; Terauchi, M.; Vikulina, T.; Roser-Page, S.; Weitzmann, M.N. B-lymphocyte, Production of both OPG and RANKL is significantly increased in aged mice. Open Bone J. 2014, 6, 8–17.
- Onal, M.; Xiong, J.; Chen, X.; Thostenson, J.D.; Almeida, M.; Manolagas, S.C.; O’Brien, C.A. Receptor activator of nuclear factor kB ligand (RANKL) protein expression by B-lymphocytes contributes to ovariectomy-induced bone loss. J. Biol. Chem. 2012, 287, 29851–29860.
- Zhang, Z.; Yuan, W.; Deng, J.; Wang, D.; Zhang, T.; Peng, L.; Tian, H.; Wang, Z.; Ma, J. Granulocyte colony stimulating factor (G-CSF) regulates neutrophils infiltration and periodontal tissue destruction in an experimental periodontitis. Mol. Immunol. 2020, 117, 110–121.
- Makiishi-Shimobayashi, C.; Tsujimura, T.; Iwasaki, T.; Yamada, N.; Sugihara, A.; Okamura, H.; Hayashi, S.; Terada, N. Interleukin-18 up-regulates osteoprotegerin expression in stromal/osteoblastic cells. Biochem. Biophys. Res. Commun. 2001, 281, 361–366.
- Makiishi-Shimobayashi, C.; Tsujimura, T.; Iwasaki, T.; Yamada, N.; Sugihara, A.; Omakura, H.; Hayashi, S.; Terada, N. IL-18BP is decreased in osteoporotic women: Prevents Inflammasome mediated IL-18 activation and reduces Th17 differentiation. Sci. Rep. 2016, 6, 33680.
- Want, R.X.; Yu, C.R.; Dambuza, I.M.; Mahdi, R.M.; Dolinska, M.B.; Sergeev, Y.V.; Wingfield, P.; Kim, S.H.; Egwuagu, C.E. Interleukin 35 induces regulator B cells that suppress autoimmune disease. Nat. Med. 2014, 20, 633–641.
- Sakkass, L.I.; Mavropoulos, A.; Perricone, C.; Bogdanos, D.P. IL-35: A new immunomodulator in autoimmune rheumatic diseases. Immunol. Res. 2018, 66, 305–312.
- Borja-Flores, F.; Bosma, A.; Ng, D.; Reddy, V.; Ehrenstein, M.R.; Isenberg, D.A.; Mauri, C. CD19+ CD24hi CD38hi B-lymphocytes maintain regulatory T-cells while limiting TH1 and TH17 differentiations. Sci. Transl. Med. 2013, 5, 173.
- Yanaba, K.; Bouaziz, J.D.; Haas, K.M.; Poe, J.C.; Fujimoto, M.; Tedder, T.F. A Regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T-cell dependent inflammatory responses. Immunity. 2008, 28, 639–650.
- Sapra, L.; Bhardwaj, A.; Mishra, P.K.; Garg, B.; Verma, B.; Mishra, G.C.; Srivastava, R.K. Regulatory B cells (Bregs) inhibit osteoclastogenesis and play a potential role in ameliorating ovariectomy-induced bone loss. Front. Immunol. 2021, 12, 691081.
- Xiong, Y.; Yan, C.; Chen, L.; Endo, Y.; Sun, Y.; Zhou, W.; Hu, Y.; Hu, L.; Chen, D.; Xue, H.; et al. IL-10 induces MC3-T3-E1 cells differentiation towards osteoblastic fate in murine model. J. Cell. Mol. Med. 2021, 24, 1076–1086.
- Evans, K.; Fox, S.W. Interleukin-10 inhibits osteoclastogenesis by reducing NFATc1 expression and preventing its translocation to the nucleus. BMC Cell Biol. 2007, 8, 4.
- Sun, G.; Wang, Y.; Ti, Y.; Wang, J.; Zhao, J.; Qian, H. Regulatory B-lymphocyte is critical in bone union process through suppressing proinflammatory cytokines and stimulating Foxp3 in Treg cells. Clin. Exp. Pharmacol. Physio. 2017, 44, 455–462.
- Lee, B.; Oh, Y.; Jo, S.; Kim, T.H.; Ji, J.D. A dual role of TGF-β in human osteoclast differentiation mediated by SMAD1 versus SMAD3 signaling. Immunol. Lett. 2019, 206, 33–40.
- Chen, G.; Deng, C.; Li, Y.P. TGF- β and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 2012, 8, 272–288.
More