Parkinson’s Disease (PD) is a prevalent neurodegenerative illness resulting in progressive motor impairment and cognitive dysfunction. Most PD cases are sporadic, and only a low percentage is related to mutations in a few genes, causing familial PD. As in other prevalent neurodegenerative disorders, aging is the principal risk factor for developing this condition. The protein cysteinome is crucial in cellular regulation and plays unexpected roles in the aging of complex organisms, which show cumulative somatic mutations, telomere attrition, epigenetic modifications, and oxidative dysregulation, culminating in cellular senescence. The cysteine thiol groups are highly redox-active, allowing high functional versatility as structural disulfides, redox-active disulfides, active-site nucleophiles, proton donors, and metal ligands to participate in multiple regulatory sites in proteins. Also, antioxidant systems control diverse cellular functions, including the transcription machinery, which partially depends on the catalytically active cysteines that can reduce disulfide bonds in numerous target proteins, driving their biological integration.
- Cysteine
- glutathione
- redox homeostasis
- NAC
- cysteinet
- Parkinson
1. Introduction
2. Role of Glutathione Precursors in Parkinson’s Disease
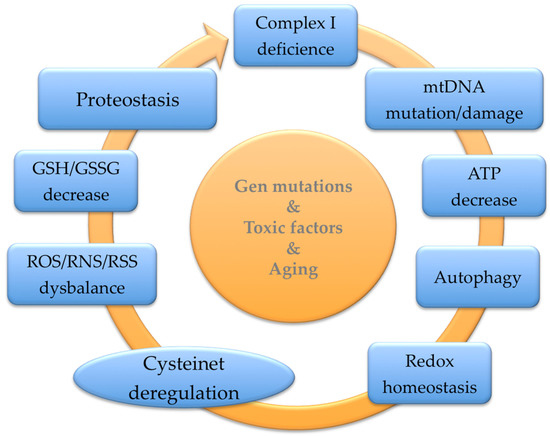
2.1. Nutrients and GSH Status
2.1.1. Amino Acids, Peptides, and Proteins
2.1.2. Vitamins
2.1.3. Flavonoids and Thiol-Rich Compounds
2.1.4. Michael Acceptor Molecules (MAMs)
2.2. CoQ10-Related Compounds
2.3. N-Acetyl-Cysteine (NAC)
2.3.1. NAC Bioavailability and Safety
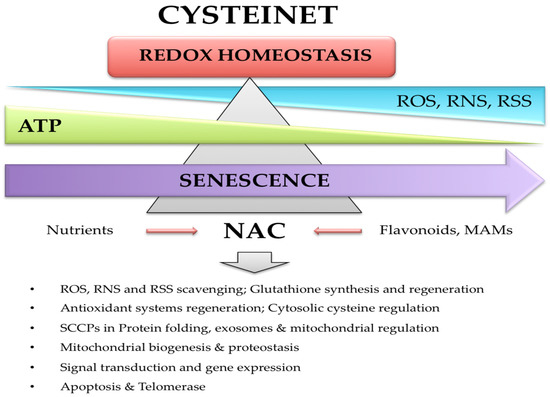
2.3.2. NAC in Cysteinet Regulation
2.3.3. NAC in Protein Misfolding
2.3.4. NAC in Cellular Vesicle Regulation and Signaling
2.3.5. NAC and Mitochondria
2.3.6. NAC and Protein Kinases
2.3.7. NAC and Telomerase
2.3.8. NAC-GSH and Nanotechnology
References
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; Collado-Mateo, D.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953.
- Veldman, B.A.; Wijn, A.M.; Knoers, N.; Praamstra, P.; Horstinka, M.W.I.M. Genetic and environmental risk factors in Parkinson’s Disease. Clin. Neurol. Neurosurg. 1998, 100, 15–26.
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888.
- Reed, X.; Bandres-Ciga, S.; Blauwendraat, C.; Cookson, M.R. The role of monogenic genes in idiopathic Parkinson’s disease. Neurobiol. Dis. 2019, 124, 230–239.
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178.
- Youdim, M.B. Understanding Parkinson’s disease. Sci. Am. 1997, 276, 52–59.
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272.
- Gelb, D.J.; Oliver, E.; Gilman, S. Diagnostic criteria for Parkinson disease. Arch. Neurol. 1999, 56, 33–39.
- Schulz-Schaeffer, W.J. The synaptic pathology of a-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143.
- Schulz-Schaeffer, W.J. Is cell death primary or secondary in the pathophysiology of idiopathic Parkinson’s disease? Biomolecules 2015, 5, 1467–1479.
- Jellinger, K.A. A critical reappraisal of current staging of Lewy related pathology in human brain. Acta Neuropathol. 2008, 116, 1–16.
- Olanow, C.W.; Perl, D.P.; DeMartino, G.N.; McNaught, K.S. Lewy body formation is an aggresome-related process: A hypothesis. Lancet Neurol. 2004, 3, 496–503.
- Fahn, S.; Cohen, G. The oxidant stress hypothesis in Parkinson’s disease: Evidence supporting it. Ann. Neurol. 1992, 32, 804–812.
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53, S26–S38.
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitocondrial dysfunction in Parkinson’s disease. Antioxid. Redox Sign. 2012, 16, 920–924.
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208.
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896.
- Lee, Z.W.; Low, Y.L.; Huang, S.; Wang, T.; Deng, L.W. The cystathionine γ-lyase/hydrogen sulfide system maintains cellular glutathione status. Biochem. J. 2014, 460, 425–435.
- Martinez-Banaclocha, M. Cellular cysteine network (Cysteinet): Pharmacological intervention in brain aging and neurodegenerative diseases. In Frontiers in Clinical Drug Research-Central Nervous System; Atta-ur-Rahman, Ed.; Bentham Science Publishers: Al Sharjah, United Arab Emirates, 2016; Volume 2, pp. 105–172.
- Lux, O.; Naidoo, D. Biological variability of superoxide dismutase and glutathione peroxidase in blood. Redox Rep. 1995, 1, 331–335.
- Tosukhowong, P.; Boonla, C.; Dissayabutra, T.; Kaewwilai, L.; Muensri, S.; Chotipanich, C.; Joutsa, J.; Rinne, J.; Bhidayasiri, R. Biochemical and clinical effects of Whey protein supplementation in Parkinson’s disease: A pilot study. J. Neurol. Sci. 2016, 367, 162–170.
- Zavorsky, G.S.; Kubow, S.; Grey, V.; Riverin, V.; Lands, L.C. An open-label dose-response study of lymphocyte glutathione levels in healthy men and women receiving pressurized whey protein isolate supplements. Int. J. Food Sci. Nutr. 2007, 58, 429–436.
- Sechi, G.; Deledda, M.G.; Bua, G.; Satta, W.M.; Deiana, G.A.; Pes, G.M.; Rosati, G. Reduced intravenous glutathione in the treatment of early Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 1996, 20, 1159–1170.
- Meister, A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacol. Ther. 1991, 51, 155–194.
- Anderson, M.E.; Meister, A. Glutathione monoesters. Anal. Biochem. 1989, 183, 16–20.
- Johnston, C.S.; Meyer, C.G.; Srilakshmi, J.C. Vitamin C elevates red blood cell glutathione in healthy adults. Am. J. Clin. Nutr. 1993, 58, 103–105.
- Sharma, A.; Kharb, S.; Chugh, S.N.; Kakkar, R.; Singh, G.P. Evaluation of oxidative stress before and after control of glycemia and after vitamin E supplementation in diabetic patients. Metabolism 2000, 49, 160–162.
- Jain, S.K.; McVie, R.; Smith, T. Vitamin E supplementation restores glutathione and malondialdehyde to normal concentrations in erythrocytes of type 1 diabetic children. Diabetes Care 2000, 23, 1389–1394.
- Ashoori, M.; Saedisomeolia, A. Riboflavin (vitamin B2) and oxidative stress: A review. Br. J. Nutr. 2014, 111, 1985–1991.
- Martins, V.D.; Manfredini, V.; Peralba, M.C.; Benfato, M.S. Alpha-lipoic acid modifies oxidative stress parameters in sickle cell trait subjects and sickle cell patients. Clin. Nutr. 2009, 28, 192–197.
- Khalili, M.; Eghtesadi, S.; Mirshafiey, A.; Eskandari, G.; Sanoobar, M.; Sahraian, M.A.; Motevalian, A.; Norouzi, A.; Moftakhar, S.; Azimi, A. Effect of lipoic acid consumption on oxidative stress among multiple sclerosis patients: A randomized controlled clinical trial. Nutr. Neurosci. 2014, 17, 16–20.
- Minich, D.M.; Brown, B.I. A review of dietary (phyto)nutrients for glutathione support. Nutrients 2019, 11, 2073.
- Yao, J.; Zhang, B.; Ge, C.; Peng, S.; Fang, J. Xanthohumol, a polyphenol chalcone present in hops, activating Nrf2 enzymes to confer protection against oxidative damage in PC12 cells. J. Agric. Food Chem. 2015, 63, 1521–1531.
- Lee, I.S.; Lim, J.; Gal, J.; Kang, J.C.; Kim, H.J.; Kang, B.Y.; Choi, H.J. Antiinflammatory activity of xanthohumol involves heme oxygenase-1 induction via NRF2-ARE signaling in microglial BV2 cells. Neurochem. Int. 2011, 58, 153–160.
- Hearn, B.R.; Fontaine, S.D.; Schneider, E.L.; Kraemer, Y.; Ashley, G.W.; Santi, D.V. Attenuation of the reaction of Michael acceptors with biologically important nucleophiles. Bioconjug. Chem. 2021, 32, 794–800.
- Liang, S.T.; Chen, C.; Chen, R.X.; Li, R.; Chen, W.L.; Jiang, G.H.; Du, L.L. Michael acceptor molecules in natural products and their mechanism of action. Front. Pharmacol. 2022, 13, 1033003.
- Cao, R.; Wang, J.; Wang, C. Zerumbone attenuates MPP+-induced cytotoxicity in human neuroblasto-ma SH-SY5Y cells by inhibition of oxidative stress. Chin. J. Pathophysiol. 2018, 34, 1061–1066.
- Peng, S.; Hou, Y.; Yao, J.; Fang, J. Activation of Nrf2-driven antioxidant enzymes by cardamonin confers neuroprotection of PC12 cells against oxidative damage. Food Funct. 2017, 8, 997–1007.
- Ji, L.; Yuan, Y.; Luo, L.; Chen, Z.; Ma, X.; Ma, Z.; Cheng, L. Physalins with antiinflammatory activity are present in Physalis alkekengi var. franchetii and can function as Michael reaction acceptors. Steroids 2012, 77, 441–447.
- Xu, Z.; Wu, G.; Li, K.; Qian, G.; Wang, X.; Chen, W. Role of PKC/MAPK/NF-kappa B signal cascade in expression of IL-1β in rat monocytes exposed to hypoxia. Chin. J. Crit. Care Med. 2008, 9, 803–807.
- Widen, J.C.; Kempema, A.M.; Baur, J.W.; Skopec, H.M.; Edwards, J.T.; Brown, T.J.; Brown, D.A.; Meece, F.A.; Harki, D.A. Helenalin analogues targeting NF-κB p65: Thiol reactivity and cellular potency studies of varied electrophiles. ChemMedChem 2018, 13, 303–311.
- Almannai, M.; El-Hattab, A.W.; Ali, M.; Soler-Alfonso, C.; Scaglia, F. Clinical trials in mitochondrial disorders, an update. Mol. Genet. Metab. 2020, 131, 1–13.
- Enns, G.M.; Kinsman, S.L.; Perlman, S.L.; Spicer, K.M.; Abdenur, J.E.; Cohen, B.H.; Amagata, A.; Barnes, A.; Kheifets, V.; Shrader, W.D.; et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol. Genet. Metab. 2012, 105, 91–102.
- Zesiewicz, T.; Salemi, J.L.; Perlman, S.; Sullivan, K.L.; Shaw, J.D.; Huang, Y.; Isaacs, C.; Gooch, C.; Lynch, D.R.; Klein, M.B. Double-blind, randomized and controlled trial of EPI-743 in Friedreich’s ataxia. Neurodegener. Dis. Manag. 2018, 8, 233–242.
- Pastore, A.; Petrillo, S.; Tozzi, G.; Carrozzo, R.; Martinelli, D.; Dionisi-Vici, C.; Di Giovamberardino, G.; Ceravolo, F.; Klein, M.B.; Miller, G.; et al. Glutathione: A redox signature in monitoring EPI-743 therapy in children with mitochondrial encephalomyopathies. Mol. Genet. Metab. 2013, 109, 208–214.
- Martinelli, D.; Catteruccia, M.; Piemonte, F.; Pastore, A.; Tozzi, G.; Dionisi-Vici, C.; Pontrelli, G.; Corsetti, T.; Livadiotti, S.; Kheifets, V.; et al. EPI-743 reverses the progression of the pediatric mitochondrial disease–genetically defined Leigh Syndrome. Mol. Genet. Metab. 2012, 107, 383–388.
- Zesiewicz, T.; Allison, K.; Jahan, I.; Shaw, J.; Murtagh, F.; Jones, T.; Gooch, C.; Salemi, J.; Klein, M.; Miller, G.; et al. EPI-743 improves motor function and CNS biomarkers in PD: Results from a phase 2A pilot trial (S40.004). Neurology 2016, 86 (Suppl. S16), I1.012.
- Martinez-Banaclocha, M. N-Acetyl-Cysteine: Modulating the cysteine redox proteome in neurodegenerative diseases. Antioxidants 2022, 11, 416.
- Martinez-Banaclocha, M. Cysteine network (CYSTEINET) dysregulation in Parkinson’s disease: Role of N-acetylcysteine. Curr. Drug Metab. 2016, 17, 368–385.
- Martinez-Banaclocha, M. N-acetylcysteine: A natural antidote for Alzheimer’s disease. Alzheimers Dis. Dement. 2016, 1, 4–15.
- Martinez-Banaclocha, M. Potential role of N-acetyl-cysteine in the cysteine proteome in Parkinson’s sisease? Clin. Pharmacol. Ther. 2020, 107, 1055.
- Martinez-Banaclocha, M. Proteomic complexity in Parkinson’s disease: A redox signaling perspective of the pathophysiology and progression. Neuroscience 2021, 453, 287–300.
- Miquel, J.; Ferrándiz, M.L.; De Juan, E.; Sevilla, I.; Martinez-Banaclocha, M. N-acetylcysteine protecs against age-related decline of oxidative phosphorylation in liver mitochondria. Eur. J. Pharmacol. 1995, 292, 333–335.
- Martinez-Banaclocha, M. N-acetyl-cysteine in schizophrenia: Potential role on the sensitive cysteine proteome. Curr. Med. Chem. 2020, 27, 6424–6439.
- Martinez-Banaclocha, M. N-acetylcysteine in psychiatric disorders: Possible role of cysteinet deregulation. Inter. Neuropsy. Dis. J. 2018, 12, 1–6.
- Medina, S.; Martinez-Banaclocha, M.; Hernanz, A. Antioxidants inhibit the human cortical neuron apoptosis induced by hydrogen peroxide, tumor necrosis factor alpha, dopamine and beta-amyloid peptide 1–42. Free Radic. Res. 2002, 36, 1179–1184.
- Martinez-Banaclocha, M. N-acetylcysteine elicited increase in complex I activity in synaptic mitochondria from aged mice: Implications for treatment of Parkinson’s disease. Brain Res. 2000, 859, 173–175.
- Martinez-Banaclocha, M. Therapeutic potential of N-acetylcysteine in age-related mitochondrial neurodegenerative diseases. Med. Hypotheses 2001, 56, 472–477.
- Martinez-Banaclocha, M. N-acetyl-cysteine in the treatment of Parkinson’s disease. What are we waiting for? Med. Hypotheses 2012, 79, 8–12.
- Martinez-Banaclocha, M.; Martínez, N. N-acetylcysteine elicited increase in cytochrome c oxidase activity in mice synaptic mitochondria. Brain Res. 1999, 842, 249–251.
- Martinez-Banaclocha, M.; Martinez, N.; Hernandez, A.I.; Ferrandiz, M.L. Hypothesis: Can N-acetylcysteine be beneficial in Parkinson’s disease? Life Sci. 1999, 64, 1253–1257.
- Martinez-Banaclocha, M. Cysteinet dysregulation in muscular dystrophies: A pathogenic network susceptible to therapy. Curr. Med. Chem. 2017, 24, 312–330.
- Martinez-Banaclocha, M.; Hernandez, A.I.; Martinez, N. N-Acetylcysteine delays age-associated memory impairment in mice: Role in synaptic mitochondria. Brain Res. 2000, 855, 100–106.
- Martinez-Banaclocha, M.; Hernandez, A.I.; Martínez, N.; Ferrándiz, M.L. N-acetylcysteine protects against age-related increase in oxidized proteins in mouse synaptic mitochondria. Brain Res. 1997, 762, 256–258.
- Miquel, J.; Martinez-Banaclocha, M.; Díez, A.; De Juan, E.; Soler, A.; Ramirez, A.; Laborda, J.; Carrión, M. Effects of turmeric on blood and liver lipoperoxide levels of mice: Lack of toxicity. Age 1995, 18, 171–174.
- Martinez-Banaclocha, M. Interfering with the reactive cysteine proteome in COVID-19. Curr. Med. Chem. 2022, 29, 1657–1663.
- Sekhar, R.V.; Patel, S.G.; Guthikonda, A.P.; Reid, M.; Balasubramanyam, A.; Taffet, G.E.; Jahoor, F. Deficient synthesis of glutathione underlies oxidative stress in aging and can be corrected by dietary cysteine and glycine supplementation. Am. J. Clin. Nutr. 2011, 94, 847–853.
- Holmay, M.J.; Terpstra, M.; Coles, L.D.; Mishra, U.; Ahlskog, M.; Oz, G.; Cloyd, J.C.; Tuite, P.J. N-Acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. Clin. Neuropharmacol. 2013, 36, 103–106.
- Caro, L.; Ghizzi, A.; Costa, R.; Longo, A.; Ventresca, G.P.; Lodola, E. Pharmacokinetics and bioavailability of oral acetylcysteine in healthy volunteers. Arzneimittelforschung 1989, 39, 382–386.
- Van Zandwijk, N. N-Acetylcysteine for lung cancer prevention. Chest 1995, 107, 1437–1441.
- De Flora, S.; Astengo, M.; Serra, D.; Bennicelli, C. Inhibition of urethan-induced lung tumors in mice by dietary N-acetylcysteine. Cancer Lett. 1986, 32, 235–241.
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta 2013, 1830, 4117–4129.
- Schwalfenberg, G.K. N-Acetylcysteine: A review of clinical usefulness (an old drug with new tricks). J. Nutr. Metab. 2021, 2021, 9949453.
- De Flora, S.; Izzotti, A.; D’Agostini, F.; Balansky, R.M. Mechanisms of N-acetylcysteine in the prevention of DNA damage and cancer, with special reference to smoking-related end-points. Carcinogenesis 2001, 22, 999–1013.
- Dekhuijzen, P.N.; van Beurden, W.J. The role for N-acetylcysteine in the management of COPD. Int. J. Chron. Obstruct. Pulmon Dis. 2006, 1, 99–106.
- Martinez-Banaclocha, M.; Hernandez, A.I.; Martinez, N.; Ferrandiz, M.L. Age-related increase in oxidized proteins in mouse synaptic mitochondria. Brain Res. 1996, 731, 246–248.
- Martinez-Banaclocha, M.; Ferrandiz, M.L.; Díez, A.; Miquel, J. Depletion of cytosolic GSH decreases the ATP levels and viability of synaptosomes from aged mice but not from young mice. Mech. Ageing Dev. 1995, 84, 77–81.
- Ferrandiz, M.L.; Martinez-Banaclocha, M.; De Juan, E.; Díez, A.; Bustos, G.; Miquel, J. Impairment of mitochondrial oxidative phosphorylation in the brain of aged mice. Brain Res. 1994, 644, 335–338.
- Martinez-Banaclocha, M.; Ferrandiz, M.L.; De Juan, E.; Miquel, J. Age-related changes in glutathione and lipid peroxide content in mouse synaptic mitochondria: Relationship to cytochrome c oxidase decline. Neurosci. Lett. 1994, 170, 121–124.
- Yan, C.Y.; Greene, L.A. Prevention of PC12 cell death by NAcetylcysteine requires activation of the Ras pathway. J. Neurosci. 1998, 18, 4042–4049.
- Hobbs, G.A.; Gunawardena, H.P.; Campbell, S.L. Biophysical and proteomic characterization strategies for cysteine modifications in Ras GTPases. Meth. Mol. Biol. 2014, 1120, 75–96.
- Tenório, M.C.D.S.; Graciliano, N.G.; Moura, F.A.; Oliveira, A.C.M.; Goulart, M.O.F. N-Acetylcysteine (NAC): Impacts on human health. Antioxidants 2021, 10, 967.
- Studer, R.; Baysang, G.; Brack, C. N-acetyl-L-cystein downregulates beta-amyloid precursor protein gene transcription in human neuroblastoma cells. Biogerontology 2001, 2, 55–60.
- García-Piñeres, A.J.; Castro, V.; Mora, G.; Schmidt, T.J.; Strunck, E.; Pahl, H.L.; Merfort, I. Cysteine 38 in p65/NF-κB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. J. Biol. Chem. 2001, 276, 39713–39720.
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Ann. Rev. Cell Dev. Biol. 2011, 27, 513–537.
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-B signaling: 785 and counting. Oncogene 2006, 25, 6887–6899.
- Yin, Z.; Machius, M.; Nestler, E.J.; Rudenko, G. Activator protein-1: Redox switch controlling structure and DNA-binding. Nucleic Acids Res. 2017, 45, 11425–11436.
- Del Sorbo, L.; Zhang, H. Is there a place for N-acetylcysteine in the treatment of septic shock? Crit. Care 2004, 8, 93–95.
- Mokhtari, V.; Afsharian, P.; Shahhoseini, M.; Kalantar, S.M.; Moini, A. A review on various uses of N-acetyl cysteine. Cell J. 2017, 19, 11–17.
- Tardiolo, G.; Bramanti, P.; Mazzon, E. Overview on the effects of N-acetylcysteine in neurodegenerative diseases. Molecules 2018, 23, 3305.
- Reynaud, E. Protein misfolding and degenerative diseases. Nat. Educ. 2010, 3, 28.
- Takai, E.; Uda, K.; Yoshida, T.; Zako, T.; Maeda, M.; Shiraki, K. Cysteine inhibits the fibrillisation and cytotoxicity of amyloid-β 40 and 42: Implications for the contribution of the thiophilic interaction. Phys. Chem. Chem. Phys. 2014, 16, 3566–3572.
- Wright, D.J.; Renoir, T.; Smith, Z.M.; Frazier, A.E.; Francis, P.S.; Thorburn, D.R.; McGee, S.L.; Hannan, A.J.; Gray, L.J. N-acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl. Psychiatry 2015, 5, e492.
- Benedikter, B.J.; Weseler, A.R.; Wouters, E.F.M.; Savelkoul, P.H.M.; Rohde, G.G.U.; Stassen, F.R.M. Redox-dependent thiol modifications: Implications for the release of extracellular vesicles. Cell. Mol. Life Sci. 2018, 75, 2321–2337.
- Mas-Bargues, C.; Sanz-Ros, J.; Romero-García, N.; Huete-Acevedo, J.; Dromant, M.; Borrás, C. Small extracellular vesicles from senescent stem cells trigger adaptive mechanisms in young stem cells by increasing antioxidant enzyme expression. Redox Biol. 2023, 62, 102668.
- Borras, C.; Mas-Bargues, C.; Sanz-Ros, J.; Román-Domínguez, A.; Gimeno-Mallench, L.; Inglés, M.; Gambini, J.; Viña, J. Extracellular vesicles and redox modulation in aging. Free Radic. Biol. Med. 2020, 149, 44–50.
- Burgoyne, R.D.; Morgan, A. Cysteine string protein (CSP) and its role in preventing neurodegeneration. Semin. Cell Dev. Biol. 2015, 40, 153–159.
- Stefanis, L. α-synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399.
- Südhof, T.C. Synaptotagmins: Why so many? J. Biol. Chem. 2002, 277, 7629–7632.
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126.
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A quantitative tissue-specific landscape of protein redox regulation during aging. Cell 2020, 180, 968–983.
- Tsai, L.H.; Delalle, I.; Caviness, V.S., Jr.; Chae, T.; Harlow, E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 1994, 371, 419–423.
- Lew, J.; Huang, Q.Q.; Qi, Z.; Winkfein, R.J.; Aebersold, R.; Hunt, T.; Wang, J.H. A brain-specific activator of cyclin-dependent kinase 5. Nature 1994, 371, 423–426.
- Ohshima, T.; Ward, J.M.; Huh, C.G.; Longenecker, G.; Pant, H.C.; Brady, R.O.; Martin, L.J.; Kulkarni, A.B. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc. Natl. Acad. Sci. USA 1996, 93, 11173–11178.
- Qu, J.; Nakamura, T.; Cao, G.; Holland, E.A.; McKercher, S.R.; Lipton, S.A. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc. Natl. Acad. Sci. USA 2011, 108, 14330–14335.
- Zhang, P.; Yu, P.C.; Tsang, A.H.; Chen, Y.; Fu, A.K.; Fu, W.Y.; Chung, K.K.; Ip, N.Y. S-nitrosylation of cyclin-dependent kinase 5 (cdk5) regulates its kinase activity and dendrite growth during neuronal development. J. Neurosci. 2010, 30, 14366–14370.
- Hu, S.Q.; Ye, J.S.; Zong, Y.Y.; Sun, C.C.; Liu, D.H.; Wu, Y.P.; Song, T.; Zhang, G.Y. S-nitrosylation of mixed lineage kinase 3 contributes to its activation after cerebral ischemia. J. Biol. Chem. 2011, 287, 2364–2377.
- Tian, H.; Zhang, Q.; Li, H.; Zhang, G. Antioxidant N-acetylcysteine and AMPA/KA receptor antagonist DNQX inhibited mixed lineage kinase-3 activation following cerebral ischemia in rat hippocampus. Neurosci. Res. 2003, 47, 47–53.
- Chen, C.H.; Li, W.; Sultana, R.; You, M.H.; Kondo, A.; Shahpasand, K.; Kim, B.M.; Luo, M.L.; Nechama, M.; Lin, Y.M.; et al. Pin1 cysteine-113 oxidation inhibits its catalytic activity and cellular function in Alzheimer’s disease. Neurobiol. Dis. 2015, 76, 13–23.
- Rangasamy, V.; Mishra, R.; Sondarva, G.; Das, S.; Lee, T.H.; Bakowska, J.C.; Tzivion, G.; Malter, J.S.; Rana, B.; Lu, K.P.; et al. Mixed-lineage kinase 3 phosphorylates prolyl-isomerase Pin1 to regulate its nuclear translocation and cellular function. Proc. Natl. Acad. Sci. USA 2012, 109, 8149–8154.
- Haendeler, J.; Hoffmann, J.; Diehl, J.F.; Vasa, M.; Spyridopoulos, I.; Zeiher, A.M.; Dimmeler, S. Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ. Res. 2004, 94, 768–775.
- Liu, J.P. Studies of the molecular mechanisms in the regulation of telomerase activity. FASEB J. 1999, 13, 2091–2104.
- Voghel, G.; Thorin-Trescases, N.; Farhat, N.; Mamarbachi, A.M.; Villeneuve, L.; Fortier, A.; Perrault, L.P.; Carrier, M.; Thorin, E. Chronic treatment with N-acetyl-cystein delays cellular senescence in endothelial cells isolated from a subgroup of atherosclerotic patients. Mech. Ageing Dev. 2008, 129, 261–270.
- Gaucher, C.; Boudier, A.; Bonetti, J.; Clarot, I.; Leroy, P.; Parent, M. Glutathione: Antioxidant properties dedicated to nanotechnologies. Antioxidants 2018, 7, 62.