Heme oxygenases (E.C. 1:14:99:33) are vital metabolic enzymes that catalyze the rate-limiting step in the degradation of heme, with the generation of carbon monoxide, biliverdin, and iron. The inducible form, heme oxygenase-1 (HO-1), is a stress protein, whose expression is responsive to a broad spectrum of adverse chemical and physical stimuli. HO-1 is known to provide cytoprotection and can exert anti-inflammatory and immunomodulatory effects in tissues, via heme removal. HO-1 is a potential therapeutic target in inflammatory diseases. The end-products of HO-1 activity, including carbon monoxide, may contribute to HO-1 mediated protection. Carbon monoxide delivery by inhalation at low concentration, as well as through application of carbon monoxide releasing molecules (CORMs), has been explored for therapeutic potential. Recently completed clinical trials have evaluated the safety and feasibility of inhaled CO as a therapy for acute and chronic lung disease,
- Carbon monoxide
- Heme oxygenase-1
- Inflammation
- Heme
- Ischemia/reperfusion
- lung disease
Thanks so much for your check. We sincerely hope you may create this entry, since you are the expert in this academic research. You can click the “submit” button to upload it after revision. We will help you layout after you submit it. Moreover, we will link your article at the entry, so more scholars and students can look through and cite it.
Definition (Draft for you)
Heme oxygenase-1 (HO-1; E.C. 1:14:99:3) is an inducible stress protein that catalyzes the oxidative conversion of heme to carbon monoxide (CO), iron, and biliverdin (BV), the latter of which is converted to bilirubin (BR) by biliverdin reductase. HO-1 has been implicated as a cytoprotectant in various models of acute organ injury and disease (i.e., lung, kidney, heart, liver). HO-1 may function as a pleiotropic modulator of inflammatory signaling, via the removal of heme, and generation of its enzymatic degradation-products.Thus, HO-1 may serve as a general therapeutic target in inflammatory disease.
1. Introduction
Heme oxygenase-1, a vital heme degradative enzyme and inducible stress protein. Heme oxygenase (HO; E.C. 1:14:99:3) is a metabolic enzyme system first described in the late 1960s which provides the rate-limiting step in the oxidative degradation of heme [1,2]. Heme catalysis by HO requires molecular oxygen (O2) and NADPH: cytochrome p-450 reductase as the electron source [1,2] (Figure 1). The HO reaction generates several products, each with biological sequelae, including carbon monoxide (CO) (which evolves from the α-methene bridge carbon of heme), the bile pigment biliverdin-IXα (BV), and free ferrous iron, Fe(II). BV is subsequently converted to its lipid soluble metabolite bilirubin-IXα (BR) by cytosolic NADPH: biliverdin reductase (BVR) [3].
Heme degradative activity is catalyzed by two major isoforms of HO, a constitutive form (heme oxygenase 2, HO-2) and an inducible form (heme oxygenase 1, HO-1), which are products of distinct genes [4,5]. The enzymatic HO substrate, heme, provides prosthetic groups for hemoglobin, the principal O2 carrier in the blood, and serves as a cofactor for mitochondrial cytochromes and other metabolic enzymes [6,7]. Heme, in free form, may also represent a hazardous pro-oxidant compound due to the presence of the central iron, a potential catalyst of harmful free radical generating processes [8]. Indeed, heme accumulation has been implicated as a pathological mediator in various inflammatory conditions such as sepsis [9–11], malaria [12], sickle cell disease [13], and atherosclerosis [14]; with its clearance by HO-1 implicated as a protective and anti-oxidative strategy in these and other diseases.
HO-1 is not only recognized as an essential metabolic enzyme, but also as a key intermediate in the stress response and in cellular adaptation to injury [15]. The upregulation of HO activity, specifically via the transcriptional regulation of HO-1, has been widely associated with cytoprotection and the protective dampening of inflammation in many cellular and preclinical models of injury [16–18].
Under conditions where HO-1 is found to be associated with cytoprotection, a plausible hypothesis is that the individual HO reaction products contribute, individually or collectively, to the implicit cytoprotective properties of HO-1 [19]. This hypothesis does not exclude activity-independent functions of the protein, such as potential intramolecular interactions resulting in the regulation of other proteins [20]. The iron release from the HO reaction drives the synthesis of ferritin, an iron sequestering macromolecule that may contribute to cytoprotection in vascular endothelium and other cell types [21]. Both BV, and its metabolite BR, are known to exert natural antioxidant properties in serum and bile [22–24]. CO evolution may contribute to the modulation of cellular function resulting in cellular adaptation, and this notion has provided a rationale for the application of pharmacological or gaseous CO to harness its therapeutic potential [25,26]. In contrast, pro-pathogenic and detrimental roles of excess HO-1 activation have also been proposed [27,28]. For example, the cytoprotective properties of HO-1 may confer a survival advantage to tumor cells, and thus contribute to cancer progression [29]. In neurodegenerative diseases, the excessive release of pro-oxidant iron by HO activity may contribute to neural injury and disease pathogenesis [30].
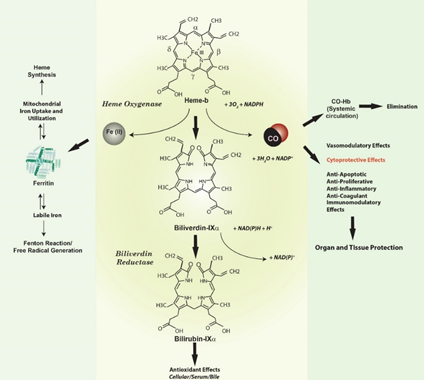
Figure 1. Heme oxygenase activity and end-product generation. Heme oxygenase (HO) is a metabolic enzyme that catalyzes the rate-limiting step in the oxidative catabolism of heme to generate the bile pigment biliverdin-IXα (BV) [1,2]. The HO reaction requires molecular oxygen (O2) and reducing equivalents from NADPH cytochrome p-450 reductase. The HO reaction releases the α-methene bridge carbon of heme as carbon monoxide (CO). The heme iron is released as ferrous iron (Fe II). BV is further metabolized to bilirubin-IXα (BR) by NAD(P)H biliverdin reductase [1–3]. Both BV and BR are potent antioxidants [22–24]. The metabolic fate of iron released by HO activity remains incompletely understood. Iron may act as a catalyst for the Haber-Weiss cycle, leading to the production of the highly reactive and deleterious hydroxyl radical, and may promote other free radical generating reactions such as the decomposition of organic peroxides [27]. Seminal studies suggested that HO-derived iron can trigger the synthesis of ferritin via modulation of iron binding protein activity [21]. Ferritin can in turn act as a potential cytoprotectant in the endothelium. Endogenously produced HO-1 derived CO has a myriad of possible biological effects, including vasoregulation, as well as cytoprotective effects that include regulation of inflammation, apoptosis, and cell proliferation, and modulation of immune responses [17,18,25,26].
The crucial role of HO-1 in systemic homeostasis, has been uncovered by rare cases of HO-1 deficiency, which have been associated with endothelial cell injury, anemia, abnormal tissue iron accumulation, inflammation, and nephritis [31,32]. HO-1 knockout mice (Hmox1-/-) show evidence of hepatic and renal iron deposition, serum anemia, and increased vulnerability to oxidative stress [33]. HO-1 is now recognized as a ubiquitous stress protein whose transcriptional regulation can be induced in cells and tissues by numerous environmental toxins and exogenous effectors [16,34]. These include both the enzyme substrate heme and its downstream catabolic product CO, physical stresses (i.e., heat shock and ultraviolet-A radiation), as well as chemical agents such as heavy metals, thiol (-SH) reactive substances, nitric oxide (NO), natural or synthetic electrophilic antioxidant compounds (e.g., curcumin, resveratrol), high and low oxygen tension (i.e., hyperoxia, hypoxia), pro-inflammatory and pro-oxidant states, pro-inflammatory cytokines, and hormones [18]. The transcriptional regulation of HO-1, while influenced by multiple nuclear factors is primarily governed by nuclear factor erythroid 2–related factor 2 (Nrf2), a master regulator of the cellular antioxidant response, which may act in combination with cell-and inducer-specific accessory transcription factors [35,36].
2. Therapeutic Modulation of HO-1 in Human Disease
The therapeutic modulation of HO-1 for amelioration of human diseases has been proposed by many investigative groups. Metalloporphyrins (i.e., tin and zinc protoporphyrin) which as competitive inhibitors of HO activity, have been extensively studied as an alternative to phototherapy for BR accumulation in neonatal jaundice [145]. More selective HO-1 inhibitors have been described [146,147]. Novel genetic knockdown methods using the CRISPR/Cas9 system have proven effective at inhibiting HO-1 expression in vitro, albeit with noted effects on cell growth, proliferation, and sensitivity to oxidative stress [148].
The prospects of gene therapy approach to drive HO-1 expression are limited by current technology advances in this domain. Since HO-1 generates several reaction products with multiple effector functions, the effects of HO-1 gene delivery will likely incur pleiotropic and off-target effects. These efforts may benefit from highly targeted cell-or tissue-specific approaches to gene delivery, as recently demonstrated in adipocytes [149]. Natural antioxidants can be used to target systemic HO-1 expression, and the mechanisms of action of many such natural plant derived substances including polyphenolic antioxidants, has been related in part to enhanced HO-1 expression [150–152]. Use of these compounds orally, will likely be non-selective. Synthetic inducers of HO-1 such as cobalt-protoporphyrin may also have pleiotropic effects and unsuitable toxicity profiles for human use/consumption. Dimethylfumarate, a drug developed for multiple sclerosis, activates Nrf2-dependent antioxidant responses, and may have further potential for clinical use [153]. Among the natural HO-1 enzymatic byproducts, iCO remains closer to therapeutic development, as discussed below.
3. Inhaled Carbon Monoxide (iCO): Summary of Current Clinical Progress
Based on a solid foundation of preclinical studies that were performed over the last two decades in rodents and higher animals (including swine and NHPs), ongoing investigations continue to seek the development of inhaled CO (iCO) as a therapeutic agent for critical care medicine applications and other human diseases. Despite initial promise of preclinical studies, therapeutic development has yet to deliver an immediate breakthrough, due to funding, regulatory hurdles, and public misconceptions despite the demonstration of safety [154,155]. Nevertheless, completed clinical trials to date have demonstrated that therapeutic application of iCO in humans is well-tolerated at low dose, and feasible [156,157]. Early trials of patients with chronic lung disease demonstrated that iCO was safe and did not cause adverse events [157]. A recent multicenter phase IIa, double-blinded, sham-controlled, clinical trial was completed, in which idiopathic pulmonary fibrosis patients were randomized to receive iCO or medical air for 12 weeks [158]. The study reported no differences in physiological measures, incidence of acute exacerbations, hospitalization, death, patient-reported outcomes, or secondary endpoints. Furthermore, no differences were reported in the distribution of adverse events. This study concluded that iCO can be safely administered to IPF patients [158].
A phase I dose escalation trial was recently performed to assess feasibility and safety of low-dose iCO administration in patients with sepsis-induced ARDS. Study subjects (n = 12) were randomized to iCO or placebo air 2:1 in two cohorts. In this trial, patients were administered iCO (100–200 ppm) or placebo air for 90 min for five days [123]. No participants exceeded a CO-Hb level of 10%, and no adverse events were reported. iCO-treated participants (100–200 ppm) displayed significant increases in COHb (up to 5%) compared with placebo-treated subjects. These studies reported that the Coburn–Forster–Kane (CFK) equation was highly accurate at predicting CO-Hb levels in patients exposed to 200 ppm CO. The study concluded that precise administration of low-dose iCO is safe and feasible in patients with sepsis-induced ARDS. Furthermore, circulating mtDNA levels were reduced in the iCO treatment groups. The study provided a precedent for ongoing Phase II studies (NCT03799874) and planned future efficacy trials [123].