Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Dean Liu and Version 3 by Dean Liu.
Some examples of carbonylative double cyclization processes, which allow the one-step synthesis of complex molecular architectures from simple building blocks using the simplest and readily available C-1 unit (CO), are illustrated and discussed.
- carbonylation
- cyclization
- double cyclization
- fused heterocycles
1. Introduction
The importance of carbon monoxide as a C-1 unit in organic synthesis can hardly be overemphasized [1]. It is a readily available feedstock that can be easily obtained by steam reforming of light hydrocarbons (including natural gas), partial oxidation of petroleum hydrocarbons, or gasification of coal to give syngas (CO and H2) [2]. It can be installed into an organic substrate, usually under catalytic conditions, leading to the direct formation of high value-added carbonylated compounds with 100% atom economy (carbonylation reactions) [1].
Here, some examples of carbonylative double cyclization processes for the synthesis of carbonylated fused heterocycles (in particular, starting from suitably functionalized olefinic substrates) are presented.
2. Functionalized Olefinic Substrates
It is well-know that palladium(II)-based catalysts activate unsaturated carbon–carbon bonds towards the attack of a variety of nucleophilic groups (mainly oxygen- or nitrogen-based). The intramolecular version of this reactivity is of particular importance as it allows the construction of heterocyclic derivatives in a straightforward manner and under mild reaction conditions (Pd(II)-catalyzed heterocyclization reactions) [3]. On the other hand, it is also very well known that Pd(II) catalysts promote many important kinds of carbonylation processes, particularly under oxidative conditions, including cyclization processes in which carbon monoxide is inserted as a carbonyl function inside the newly formed ring (cyclocarbonylation reactions) [4]. It is therefore not surprising that several important methods have been developed in which a single Pd(II)-based catalytic system promotes, in one synthetic step, the sequential heterocyclization−cyclocarbonylation of suitably functionalized olefinic substrates that carry two nucleophilic moieties placed in appropriate positions to undergo double cyclization. Pioneering studies on this kind of reactivity were conducted by the Semmelhack and Yoshida research groups during the 1980s. In 1984, Semmelhack et al. reported the Pd(II)-promoted stereoselective carbonylative double cyclization of 1-(2-(hydroxymethyl)phenyl)prop-2-en-1-ols to give 3,3a,5,9b-tetrahydro-2H-furo[3,2-c]isochromen-2-ones with a cis junction between the newly formed rings using a stoichiometric amount of Pd(OAc)2 [5]. The process started with the intramolecular 6-exo-trig nucleophilic attack of the benzylic hydroxyl group to the double bond, activated by coordination to the Pd(II) center. This led to the formation of a cis-type alkylpalladium intermediate stabilized by chelation of the second hydroxyl group. The final bicyclic product was then formed through CO migratory insertion followed by intramolecular nucleophilic displacement by the hydroxyl, possibly via the formation of a palladacycle followed by reductive elimination (Scheme 1).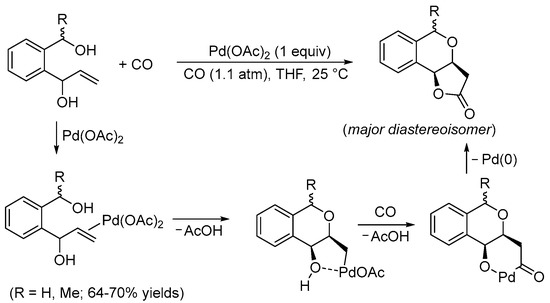
Scheme 1. Synthesis of 3,3a,5,9b-tetrahydro-2H-furo[3,2-c]isochromen-2-ones from 1-(2-(hydroxymethyl)phenyl)prop-2-en-1-ols [5].
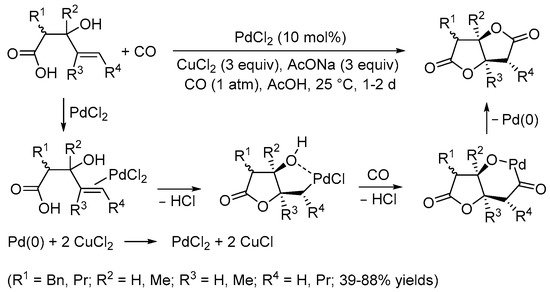
Scheme 2. Synthesis of tetrahydrofuro[3,2-b]furan-2,5-diones from 3-hydroxy-4-pentenoic acids [6].
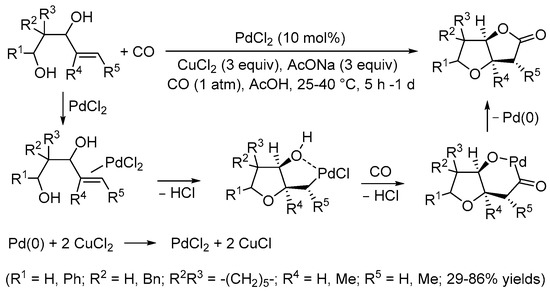
Table 1. Representative examples of the Pd(II)-promoted carbonylative double cyclization of enediol derivatives in the synthesis of natural and bioactive products.
| Entry | Conditions | Substrate | Product | Yield (%) | Refs. |
|---|---|---|---|---|---|
| 1 | PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 41 h |  |
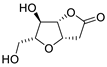 |
63 | [8] |
| 2 | PdCl2(MeCN)2, (10 mol%), CuCl2 (2.4 equiv), CO (1 atm), THF, 25 °C, 24 h | 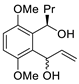 |
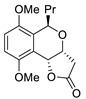 |
65 | [9] |
| 3 | PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 8 h | 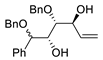 |
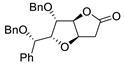 |
85 | [10] |
| 4 | PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (4 equiv), CO (1 atm), AcOH, 25 °C, 24 h | 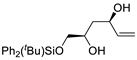 |
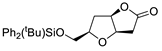 |
93 | [11] |
| 5 | PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 33 h | 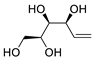 |
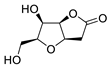 |
38 | [12] |
| 6 | PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 15 h | 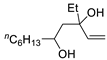 |
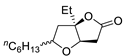 |
>80 | [13] |
| 7 | Pd(OAc)2 (1.5 equiv), CO (1.1 atm), THF, 23 °C, 4 h |
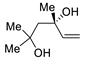 |
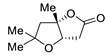 |
87 | [14] |
| 8 | PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 15 h | 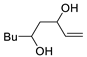 |
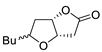 |
81 | [15] |
| 9 | PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C | 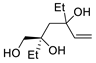 |
 |
63 | [16] |
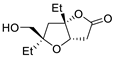
| 10 | PdCl2, CuCl, AcONa, CO, AcOH | 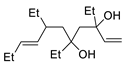 |
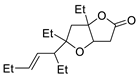 |
33 | [17] |
| 11 | PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 10 h | 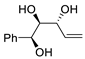 |
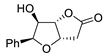 |
85 | [18] |
| 12 | PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 23 °C, 24 h | 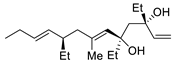 |
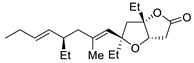 |
75 | [19] |
| 13 | Pd(OAc)2 (1.5 equiv), N-methylmorpholine (3 equiv), CO, THF, 25 °C, 15 h |
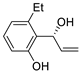 |
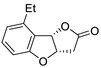 |
58 | [20] |
| 14 | PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 20 h | 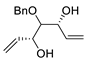 |
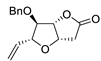 |
65 | [21] |
| 15 | Pd(OAc)2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 15 h |
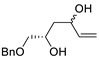 |
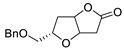 |
63, 70 | [22][23] |
| 16 | PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 24 h | 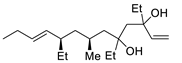 |
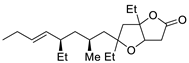 |
33 | [24] |
| 17 | PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 24 h | 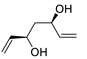 |
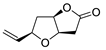 |
87 | [25] |
| 18 | PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 12 h | 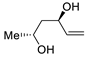 |
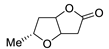 |
61 | [26] |
| 19 | PdCl2(MeCN)2 (10 mol%), CuCl2 (5 equiv), AcOLi (5 equiv), [Fe(CO)5] (0.5 equiv), AcOH, 60 °C, 1 h | 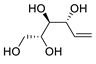 |
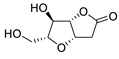 |
47 | [27] |
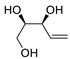
| 20 | PdCl2(MeCN)2 (10 mol%), Cu(OAc)2 (4 equiv), LiCl (4 equiv), [Fe(CO)5] (0.25 equiv), AcOH, 60 °C, 15 min |
 |
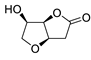 |
67 | [28] |
| 21 | PdCl2(MeCN)2 (10 mol%), CuCl2 (4 equiv), AcOLi (4 equiv), [Fe(CO)5] (0.3 equiv), AcOH, 60 °C, 30 min | 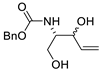 |
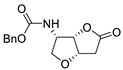 |
75 | [29] |
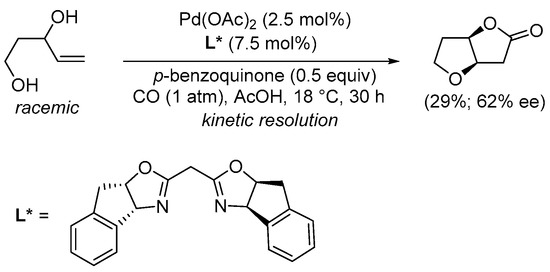
Scheme 4. Kinetic resolution of (±)-pent-4-ene-1,3-diol leading to enantioenriched (3aR,6aR)-tetrahydrofuro[3,2-b]furan-2(3H)-one [30].
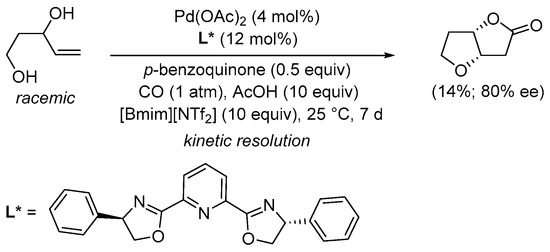
Scheme 5. Kinetic resolution of (±)-pent-4-ene-1,3-diol in [bmim][NTf2] leading to enantioenriched (3aS,6aS)-tetrahydrofuro[3,2-b]furan-2(3H)-one [31].
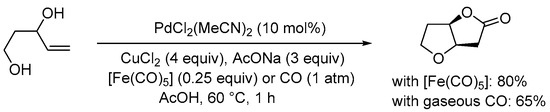
Scheme 6. Carbonylative double cyclization of pent-4-ene-1,3-diol using [Fe(CO)5] as in situ CO source [32].
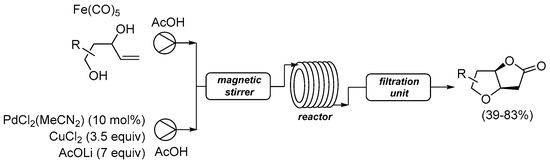
Scheme 7. Carbonylative double cyclization of 4-ene-1,3-diols using [Fe(CO)5] as in situ CO source under flow conditions [34].
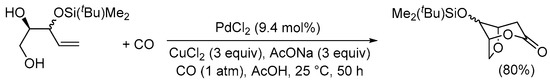
Scheme 8. 5-exo-trig O-cyclization followed by cyclocarbonylation with 6-membered ring closure [35].
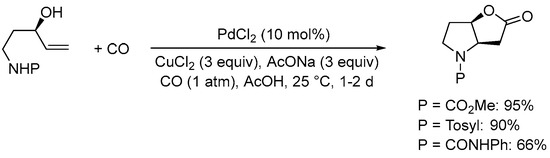
Scheme 9. Formation of 6-hydroxyhexahydro-2H-furo[3,2-b]pyrrol-2-one derivatives from N-protected 5-aminopent-1-en-3-ols [37].
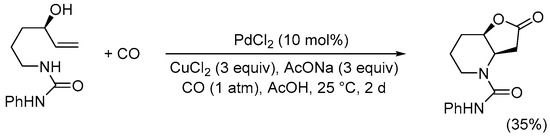
Scheme 10. Synthesis of 2-oxo-N-phenylhexahydrofuro[3,2-b]pyridine-4(2H)-carboxamide from 1-(4-hydroxyhex-5-en-1-yl)-3-phenylurea [37].
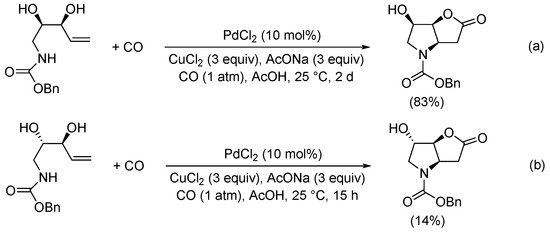
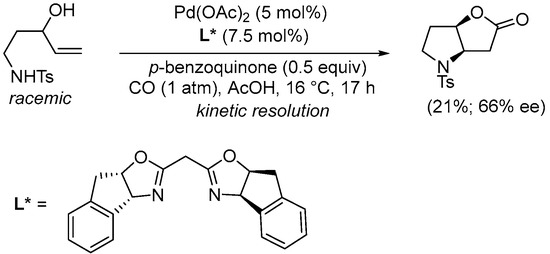
Scheme 12. Kinetic resolution of (±)-N-(3-hydroxypent-4-en-1-yl)-4-methylbenzenesulfonamide leading to enantioenriched (3aR,6aR)-4-tosylhexahydro-2H-furo[3,2-b]pyrrol-2-one [40].
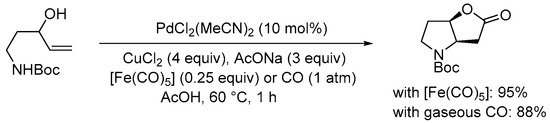
Scheme 13. Carbonylative double cyclization of tert-butyl (3-hydroxypent-4-en-1-yl)carbamate using [Fe(CO)5] as in situ CO source [32].
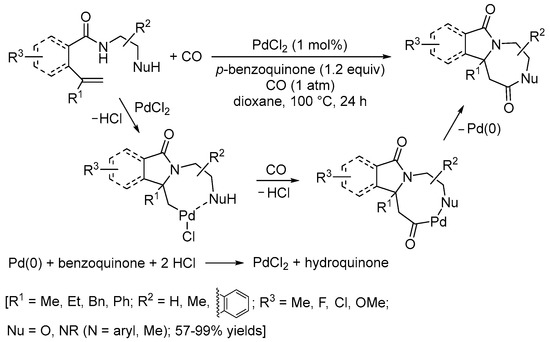
Scheme 14. Carbonylative double cyclization of N-(2-aminoethyl)pent-4-enamide and N-(2-hydroxyethyl)pent-4-enamide derivatives [41].
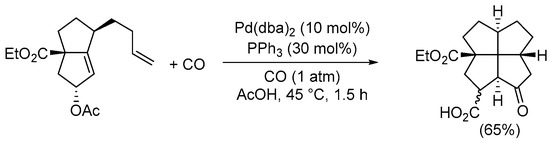
Scheme 15. Synthesis of 2a-(ethoxycarbonyl)-8-oxododecahydropentaleno[1,6-cd]pentalene-1-carboxylic acid from ethyl 5-acetoxy-1-(but-3-en-1-yl)-2,3,4,5-tetrahydropentalene-3a(1H)-carboxylate by Pauson–Khand-type intramolecular reaction [48].
References
- Gabriele, B. (Ed.) Carbon Monoxide in Organic Synthesis—Carbonylation Chemistry; Wiley-VCH: Weinheim, Germany, 2022.
- Reimert, R.; Marschner, F.; Renner, H.-J.; Boll, W.; Supp, E.; Brejc, M.; Liebner, W.; Schaub, G. Gas production, 2. In Ullmann’s Encyclopedia of Industrial Chemistry; Baltes, H., Göpel, W., Hesse, J., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp. 423–479.
- Li, J.J.; Gribble, G. (Eds.) Palladium in Heterocyclic Chemistry—A Guide for the Synthetic Chemist, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2006.
- Gabriele, B.; Della Ca’, N.; Mancuso, R.; Veltri, L.; Ziccarelli, I. Palladium(II)-Catalyzed Carbonylations. In Carbon Monoxide in Organic Synthesis—Carbonylation Chemistry; Gabriele, B., Ed.; Wiley-VCH: Weinheim, Germany, 2022; Chapter 7.
- Semmelhack, M.F.; Bodurow, C.; Baum, M. Direct Synthesis of Pyran-Lactones Related to Nafhthoquinone Antibiotics. Tetrahadron Lett. 1984, 25, 3171–3174.
- Tamaru, H.; Higashimura, K.; Hojo, M.; Yoshida, Z. PdII-Catalyzed Stereoselective Bis-Lactonization. Angew. Chem. Int. Ed. 1985, 24, 1045–1046.
- Tamaru, H.; Kobayashi, T.; Kawamura, S.; Hojo, M.; Yoshida, Z. Palladium Catalyzed Oxycarbonylation of 4-Penten-1,3-diols: Efficient Stereoselective Synthesis of cis 3-Hydroxytetrahydrofuran 2-Acetic Acid Lactones. Tetrahedron Lett. 1985, 26, 3207–3210.
- Gracza, T.; Hasenöhrl, T.; Stahl, T.; Jäger, V. Synthesis of 3,5-Anhydro-2-deoxy-1,4-glyconolactones by Palladium(II)-Catalyzed, Regioselective Oxycarbonylation of C5- and C6-enitols. ω-Homologation of Aldoses to Produce Intermediates for C-Glycoside/C-Nucleoside Synthesis. Synthesis 1991, 1991, 1108–1118.
- Kraus, G.A.; Li, J. Regiocontrol by Remote Substituents. An Enantioselective Total Synthesis of Frenolicin B via a Highly Regioselective Diels-Alder Reaction. J. Am. Chem. Soc. 1993, 115, 5859–5860.
- Gracza, T.; Jäger, V. Synthesis of Natural and Unnatural Enantiomers of Goniofufurone and Its 7-Epimers from D-Glucose. Application of Palladium(II)—Catalyzed Oxycarbonylation of Unsaturated Polyols. Synthesis 1994, 1994, 1359–1368.
- Boukouvalas, J.; Fortier, G.; Radu, I.-I. Efficient Synthesis of (−)-trans-Kumausyne via Tandem Intramolecular Alkoxycarbonylation-Lactonization. J. Org. Chem. 1998, 63, 916–917.
- Dixon, D.J.; Ley, S.V.; Gracza, T.; Szolcsanyi, P. Total Synthesis of the Polyenoyltetramic acid Mycotoxin Erythroskyrine. J. Chem. Soc. Perkin Trans. 1 1999, 1999, 831–841.
- Paddon-Jones, G.C.; Hungerford, N.L.; Haynes, P.; Kitching, W. Efficient Palladium(II)-Mediated Construction of Functionalized Plakortone Cores. Org. Lett. 1999, 1, 1905–1908.
- Semmelhack, M.F.; Shanmugam, P. Development of an Approach to the Synthesis of the Plakortones. Tetrahedron Lett. 2000, 41, 3567–3571.
- Paddon-Jones, G.C.; McErlean, C.S.P.; Haynes, P.; Moore, C.J.; Konig, W.A.; Kitching, W. Synthesis and Stereochemistry of Some Bicyclic γ-Lactones from Parasitic Wasps (Hymenoptera: Braconidae). Utility of Hydrolytic Kinetic Resolution of Epoxides and Palladium(II)-Catalyzed Hydroxycyclization-Carbonylation-Lactonization of Ene-diols. J. Org. Chem. 2001, 66, 7487–7495.
- Haynes, P.Y.; Kitching, W. Total Synthesis and Absolute Stereochemistry of Plakortone D. J. Am. Chem. Soc. 2002, 124, 9718–9719.
- Haynes, P.Y.; Kitching, W. Synthesis of the Plakortone Series: Plakortone E. Heterocycles 2004, 62, 173–177.
- Babjak, M.; Kapitán, P.; Gracza, T. Synthesis of (+)-Goniothalesdiol and (+)-7-epi-Goniothalesdiol. Tetrahedron 2005, 61, 2471–2479.
- Semmelhack, M.F.; Hooley, R.J.; Kraml, C.M. Synthesis of Plakortone B and Analogs. Org. Lett. 2006, 8, 5203–5206.
- Boukouvalas, J.; Pouliot, M.; Robichaud, J.; MacNeil, S.; Snieckus, V. Asymmetric Total Synthesis of (−)-Panacene and Correction of Its Relative Configuration. Org. Lett. 2006, 8, 3597–3599.
- Kapitán, P.; Gracza, T. Stereocontrolled Oxycarbonylation of 4-Benzyloxyhepta-1,6-diene-3,5-diols Promoted by Chiral Palladium(II) Complexes. Tetrahedron Asymm. 2008, 19, 38–44.
- Nesbitt, C.L.; McErlean, C.S.P. An Expedient Synthesis of 2,5-Disubstituted-3-oxygenated Tetrahydrofurans. Tetrahedron Lett. 2009, 50, 6318–6320.
- Nesbitt, C.L.; McErlean, C.S.P. Total Synthesis of C19 Lipid Diols Containing a 2,5-Disubstituted-3-Oxygenated Tetrahydrofuran. Org. Biomol. Chem. 2011, 9, 2198–2208.
- Haynes, P.Y.; Chow, S.; Rahm, F.; Bernhardt, P.V.; De Voss, J.J.; Kitching, W. Synthesis of the Sponge-Derived Plakortone Series of Bioactive Compounds. J. Org. Chem. 2010, 75, 6489–6501.
- Werness, J.B.; Tang, W. Stereoselective Total Synthesis of (−)-Kumausallene. Org. Lett. 2011, 13, 3664–3666.
- Markovič, M.; Ďuranová, M.; Koóš, P.; Szolcsányi, P.; Gracza, T. Synthesis of bis-Tetrahydrofuran Subunit of (−)-Neopallavicinin. Tetrahedron 2013, 69, 4185–4189.
- Markovič, M.; Koóš, P.; Čarný, T.; Sokoliová, S.; Bohačiková, N.; Moncol’, J.; Gracza, T. Total Synthesis, Configuration Assignment, and Cytotoxic Activity Evaluation of Protulactone A. J. Nat. Prod. 2017, 80, 1631–1638.
- Markovič, M.; Koóš, P.; Gracza, T. A Short Asymmetric Synthesis of Sauropunols A–D. Synthesis 2017, 49, 2939–2942.
- Lopatka, P.; Gavenda, M.; Markovič, M.; Koóš, P.; Gracza, T. Flow Pd(II)-Catalyzed Cyclisation in the Total Synthesis of Jaspine B. Catalysts 2021, 11, 1513.
- Kapitán, P.; Gracza, T. Asymmetric Intramolecular Pd(II)-catalyzed Oxycarbonylation of Alkene-1,3-diols. Arkivoc 2008, viii, 8–17.
- Doháňošová, J.; Lásikivá, A.; Toffano, M.; Gracza, T.; Vo-Thanh, G. Kinetic Resolution of Pent-4-ene-1,3-diol by Pd(II)-Catalysed Oxycarbonylation in Ionic Liquids. New J. Chem. 2012, 36, 1744–1750.
- Babjak, M.; Markovič, K.; Kandríkova, B.; Gracza, T. Homogeneous Cyclocarbonylation of Alkenols with Iron Pentacarbonyl. Synthesis 2014, 46, 809–816.
- Markovič, K.; Lopatka, P.; Koóš, P.; Gracza, T. Asymmetric Formal Synthesis of (+)-Pyrenolide D. Synthesis 2014, 46, 817–821.
- Lopatka, P.; Markovič, K.; Koóš, P.; Ley, S.V.; Gracza, T. Continuous Pd-Catalyzed Carbonylative Cyclization Using Iron Pentacarbonyl as a CO Source. J. Org. Chem. 2019, 84, 14394–14406.
- Babjak, M.; Zálupský, P.; Gracza, T. Regiocontrol in the Palladium(II)-Catalysed Oxycarbonylation of Unsaturated Polyols. Arkivoc 2005, 45, 57.
- Tamaru, Y.; Kobayashi, T.; Kawamura, S.; Ochiai, H.; Yoshida, Z. Stereoselective Intramolecular Aminocarbonylation of 3-Hydroxypent-4-enylamides Catalyzed by Palladium. Tetrahedron Lett. 1985, 26, 4479–4482.
- Tamaru, Y.; Hojo, M.; Yoshida, Z. Palladium(2+)-Catalyzed Intramolecular Aminocarbonylation of 3-Hydroxy-4-pentenylamines and 4-Hydroxy-5-hexenylamines. J. Org. Chem. 1988, 53, 5731–5741.
- Hümmer, W.; Dubois, E.; Gracza, T.; Jäger, V. Halocyclization and Palladium(II)-Catalyzed Amidocarbonylation of Unsaturated Aminopolyols. Synthesis of 1,4-Iminoglycitols as Potential Glycosidase Inhibitors. Synthesis 1997, 1997, 634–642.
- Caletková, O.; Ďurišová, N.; Gracza, T. Aminohydroxylation of Divinylcarbinol and its Application to the Synthesis of Bicyclic hydroxypyrrolidine and Aminotetrahydrofuran Building Blocks. Chem. Pap. 2013, 67, 66–75.
- Koóš, P.; Špánik, I.; Gracza, T. Asymmetric Intramolecular Pd(II)-Catalysed Amidocarbonylation of Unsaturated Amino Alcohols. Tetrahedron Asymm. 2009, 20, 2720–2723.
- Shi, L.; Weng, M.; Li, F. Palladium-Catalyzed Tandem Carbonylative Aza-Wacker-Type Cyclization of Nucleophile Tethered Alkene to Access Fused N-Heterocycles. Chin. J. Chem. 2021, 39, 317–322.
- Lee, H.-W.; Kwong, F.-Y. A Decade of Advancements in Pauson-Khand-Type Reactions. Eur. J. Org. Chem. 2010, 2010, 789–811.
- Shibata, T. Recent Advances in the Catalytic Pauson-Khand-type Reaction. Adv. Synth. Catal. 2006, 348, 2328–2336.
- Blanco-Urgoiti, J.; Añorbe, L.; Pérez-Serrano, L.; Domínguez, G.; Pérez-Castells, J. The Pauson–Khand Reaction, a Powerful Synthetic Tool for the Synthesis of Complex Molecules. Chem. Soc. Rev. 2004, 33, 32–42.
- Heravi, M.M.; Mohammadi, L. Application of Pauson-Khand Reaction in the Total Synthesis of Terpenes. RSC Adv. 2021, 11, 38325–38373.
- Yang, Z. Navigating the Pauson-Khand Reaction in Total Syntheses of Complex Natural Products. Acc. Chem. Res. 2021, 54, 556–568.
- Chen, S.; Jiang, C.; Zheng, N.; Yang, Z.; Shi, L. Evolution of Pauson-Khand Reaction: Strategic Applications in Total Syntheses of Architecturally Complex Natural Products (2016–2020). Catalysts 2020, 10, 1199.
- Keese, F.; Guidetti-Grept, R.; Herzog, B. Synthesis of Fenestranes by Pd-Catalyzed Carbonylation-Cyclisation. Tetrahedron Lett. 1992, 33, 1207–1210.
More