Nowadays, an increasing number of heterocyclic-based drugs found application in medicinal chemistry and, in particular, as anticancer agents. In this context, oxadiazoles—five-membered aromatic rings—emerged for their interesting biological properties
- bioactive heterocyclics
- anticancer agents
1. Introduction
Oxadiazoles are small five-membered heterocycles, composed of two carbon, one oxygen, and two nitrogen atoms, which attracted a lot of interest in different scientific disciplines: from medicine [1][2] and agrochemistry [3] to materials science [4][5].
Oxadiazoles are small five-membered heterocycles, composed of two carbon, one oxygen, and two nitrogen atoms, which attracted a lot of interest in different scientific disciplines: from medicine [2,3] and agrochemistry [4] to materials science [5,6].
Their aromatic flat surface is effective in the target binding, through π-stacking interactions, or to properly outdistance the substituents according to a specific orientation [6]. Depending on nitrogen atoms position, oxadiazoles exist in four different regioisomeric forms (
Their aromatic flat surface is effective in the target binding, through π-stacking interactions, or to properly outdistance the substituents according to a specific orientation [7]. Depending on nitrogen atoms position, oxadiazoles exist in four different regioisomeric forms (
): 1,2,3-, 1,2,4-, 1,2,5-, and 1,3,4-oxadiazoles.

Figure 1.
Among the four isomers, 1,2,4- and 1,3,4-oxadiazoles frequently occur in a large series of drug-like molecules [7], including antiviral [2][8], antihypertensive [9], anti-diabetic [10], anti-inflammatory, and analgesic [11], as well as anticancer compounds [12][13]. Probably, this is a direct consequence of the orientation of the side chains (-R
Among the four isomers, 1,2,4- and 1,3,4-oxadiazoles frequently occur in a large series of drug-like molecules [8], including antiviral [3,9], antihypertensive [10], anti-diabetic [11], anti-inflammatory, and analgesic [12], as well as anticancer compounds [13,14]. Probably, this is a direct consequence of the orientation of the side chains (-R
1
and -R
2
in
Figure 1), very similar for 1,2,4- and 1,3,4-oxadiazoles with respect to 1,2,3- and 1,2,5-oxadiazoles. Indeed, small bioactive molecules with resembling shapes usually show target-binding similarity that translates into the same biological effects [14]. Moreover, their bioisosteric correspondence with ester and amide groups makes oxadiazoles an interesting synthetic alternative to avoid intrinsic molecular instability (e.g., due to hydrolysis reactions), guaranteeing different hydrogen-bonding potentials with the receptors [7].
), very similar for 1,2,4- and 1,3,4-oxadiazoles with respect to 1,2,3- and 1,2,5-oxadiazoles. Indeed, small bioactive molecules with resembling shapes usually show target-binding similarity that translates into the same biological effects [15]. Moreover, their bioisosteric correspondence with ester and amide groups makes oxadiazoles an interesting synthetic alternative to avoid intrinsic molecular instability (e.g., due to hydrolysis reactions), guaranteeing different hydrogen-bonding potentials with the receptors [8].
The extensively use of this class of heterocyclic compounds in medicinal chemistry is confirmed by the presence of several commercially available drugs based on these interesting scaffolds. The reasons for their success lie in an efficient and simple synthesis, high versatility, giving rise to elevated structural diversity, and remarkable stability, a key feature for in vivo applications. Thanks to its structural nature, oxadiazoles easily interact with bio-targets establishing π-stacking interactions or forming strong hydrogen bonds.
During the last decades, the design of new oxadiazole-based scaffolds accelerated in medicinal chemistry, bringing most of these compounds to the preclinical stage or, even, to commercialization. Among these, the most commercially available drugs are Oxolamine, a cough suppressant [15], Ataluren, indicated for the treatment of Duchenne muscular dystrophy [16] and cystic fibrosis [17], Butalamine, a vasodilator, Proxazole, a drug for functional gastrointestinal disorders, Fasiplon, an anxiolytic drug [18], Raltegravir, an antiretroviral drug used to treat HIV [19], which has been recently proposed as repurposing drug against SARS-COV-2 [20], as well as the antiviral Pleconaril selected for the SARS-COV-2 spike protein [21] (
During the last decades, the design of new oxadiazole-based scaffolds accelerated in medicinal chemistry, bringing most of these compounds to the preclinical stage or, even, to commercialization. Among these, the most commercially available drugs are Oxolamine, a cough suppressant [16], Ataluren, indicated for the treatment of Duchenne muscular dystrophy [17] and cystic fibrosis [18], Butalamine, a vasodilator, Proxazole, a drug for functional gastrointestinal disorders, Fasiplon, an anxiolytic drug [19], Raltegravir, an antiretroviral drug used to treat HIV [20], which has been recently proposed as repurposing drug against SARS-COV-2 [21], as well as the antiviral Pleconaril selected for the SARS-COV-2 spike protein [22] (
). Equally well known is Zibotentan, an anticancer drug in late-stage clinical trials (
Identifier: NCT00554229), which is a candidate in development by AstraZeneca (
).
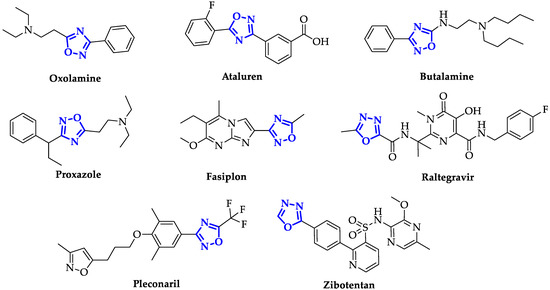
Figure 2. Structures of some of the most famous ligands bearing oxadiazole moiety.
Considering the meaningful social impact of cancer, due to the incessant raise incidence, with resistance towards existing drugs and severe side effects, the need to develop novel anticancer drugs is compelling.
Recently, it has been demonstrated that oxadiazoles can interact with specific targets, as demonstrated for enzymes, globular proteins and DNA structures. This correspondence helped to clarify the origin of cytotoxicity of these compounds, allowing the improvement of drug design in relationship with the identified target.
It should be underlined that oxadiazoles that present relevant cytotoxicity are characterized by some common structural features: they present modification with bioactive compounds, an extended aromatic surface, or the presence of a sulphur-based functional group. These results evidence the important progresses achieved in the last few years, however, a lot of effort should be dedicatedto further optimize these chemical structures in order to enhance their potency and to improve the selectivity towards specific targets.
2. Oxadiazoles Derivatives as Anticancer Agents without a Specific Target Identified
A broad number of oxadiazole ligands with promising anticancer activity have been developed despite their mechanism of action must be still clarified. In this sense, modification of oxadiazole scaffolds through conjugation to bioactive natural products represents a very promising approach. Mironov et al. exploited Furano-diterpenoids of the labdane series, an anti-inflammatory and anti-allergic drug-like [22], to build novel and potent derivatives [23]. In detail, 1,2,4-oxadiazoles were conjugated to labda-8(9),13,15-triene or labda-8(17),13,15-triene core and their antiproliferative activity and GI
A broad number of oxadiazole ligands with promising anticancer activity have been developed despite their mechanism of action must be still clarified. In this sense, modification of oxadiazole scaffolds through conjugation to bioactive natural products represents a very promising approach. Mironov et al. exploited Furano-diterpenoids of the labdane series, an anti-inflammatory and anti-allergic drug-like [90], to build novel and potent derivatives [91]. In detail, 1,2,4-oxadiazoles were conjugated to labda-8(9),13,15-triene or labda-8(17),13,15-triene core and their antiproliferative activity and GI
50 on CEM-13, MT-4 and U937 cells were measured [23]. From this screening, compound
on CEM-13, MT-4 and U937 cells were measured [91]. From this screening, compound
55
(
Figure 3) emerged for its remarkable toxicity, with GI
10) emerged for its remarkable toxicity, with GI
50
values of 0.08 ± 0.03 µM on CEM-13, and 0.35 ± 0.11 µM on MT-4. Moreover, cytofluorimetric analysis demonstrated that treatment of U937 cells with
55 induced apoptosis in 46.8% of cells after 24 h and 84.4% after 48 h [23].
induced apoptosis in 46.8% of cells after 24 h and 84.4% after 48 h [91].
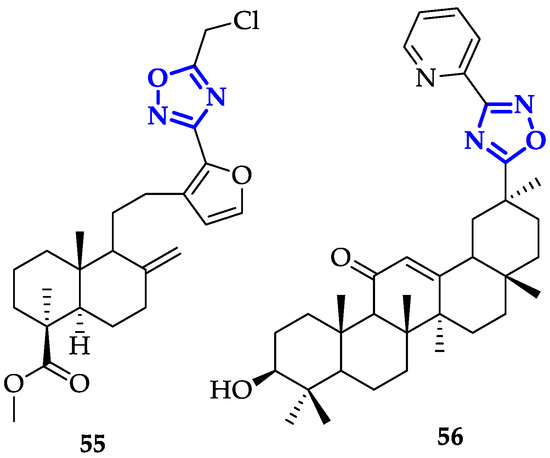
Figure 310.
Each molecule has been screened at a single dose of 100 µM against a panel of 60 cancer cell lines. From this screening
57
(
Figure 4), emerged as the most efficient antiproliferative agent, highlighting that an extended aromatic surface (two phenyl rings with electron-donating substituents) could guarantee better anticancer activity.
11), emerged as the most efficient antiproliferative agent, highlighting that an extended aromatic surface (two phenyl rings with electron-donating substituents) could guarantee better anticancer activity.
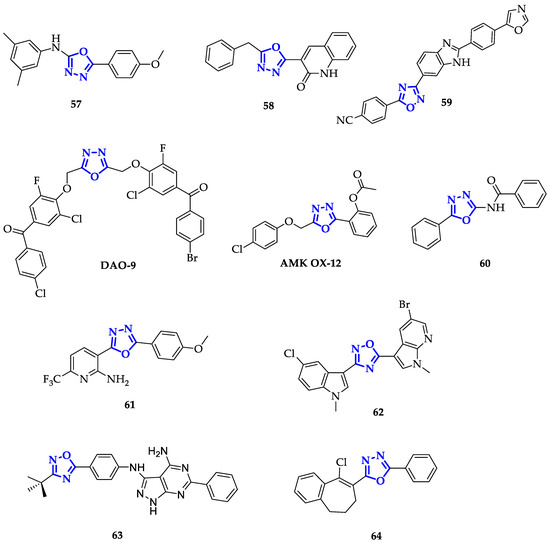
Figure 411.
Following the same approach, Salahuddin and co-workers synthesized a library of 1,3,4-oxiadiazoles conjugated to quinolines and benzimidazole derivatives and measured their growth inhibition percentage (GP) towards a broad panel of cancer cell lines [24]. Compound
Following the same approach, Salahuddin and co-workers synthesized a library of 1,3,4-oxiadiazoles conjugated to quinolines and benzimidazole derivatives and measured their growth inhibition percentage (GP) towards a broad panel of cancer cell lines [96]. Compound
58
(
Figure 4) emerged for its remarkable toxicity on HTC-116, with GP equal to 7.52 µM and GI
11) emerged for its remarkable toxicity on HTC-116, with GP equal to 7.52 µM and GI
50
of 1.41 µM. Therefore, the introduction of quinoline moiety resulted in a promising path for the development of new anticancer drugs.
Conjugation to benzimidazole scaffold has been also exploited by Srinivas et al., obtaining compound
59
(
Figure 4), highly toxic on A549 cells, with IC
11), highly toxic on A549 cells, with IC
50 of 0.12 ± 0.01 µM [25].
of 0.12 ± 0.01 µM [97].
Modification of the 1,3,4-oxadiazole scaffold with only two phenyl rings resulted to be a successful strategy also for Mochona et al., that individuated compound
60
(
Figure 4) as the best of the series with the highest toxicity in particular on PC-3 cells (IC
11) as the best of the series with the highest toxicity in particular on PC-3 cells (IC
50 = 0.22 µM) [26]. Subsequently, it has been verified that
= 0.22 µM) [101]. Subsequently, it has been verified that
60
induced apoptosis and cell cycle arrest at G0/G1 and S phases.
Introduction of 2-substituted pyridines, as reported by Kumar et al., led to promising results as demonstrated by proving the efficiency of these compounds against HeLa, DU145, HepG2, and MDA-MB-231 cancer cell lines [27]. Among them,
Introduction of 2-substituted pyridines, as reported by Kumar et al., led to promising results as demonstrated by proving the efficiency of these compounds against HeLa, DU145, HepG2, and MDA-MB-231 cancer cell lines [102]. Among them,
61
(
Figure 4) showed higher toxicity in particular on DU145 cells with an IC
11) showed higher toxicity in particular on DU145 cells with an IC
50 value of 10.7 µM. This enhanced activity has been attributed to both the presence of a trifluoromethyl group, that improves lipophilicity, and to the electron-donating effect of methoxy substituent [27].
value of 10.7 µM. This enhanced activity has been attributed to both the presence of a trifluoromethyl group, that improves lipophilicity, and to the electron-donating effect of methoxy substituent [102].
On the contrary, Cascioferro et al. extended the aromatic system of 1,2,4-oxadiazole exploiting indole and azaindole derivatives with different types of substituents, including halogen atoms and methoxy units [
]. After a preliminary screening on HCT-116 cells, compound
62
(
Figure 4) emerged as the most potent, displaying high toxicity on different cell lines and specifically on MCF-7 cells with IC
11) emerged as the most potent, displaying high toxicity on different cell lines and specifically on MCF-7 cells with IC
50
of 0.65 ± 0.05 µM. Furthermore, authors demonstrated that
62 caused cell cycle arrest (more than 60%) at G0/G1 phase and its ability to induce apoptosis was confirmed through Annexin V/PI dual staining method, followed by cytofluorimetric analysis [28].
caused cell cycle arrest (more than 60%) at G0/G1 phase and its ability to induce apoptosis was confirmed through Annexin V/PI dual staining method, followed by cytofluorimetric analysis [103].
The presence of an aromatic system is not the only important structural feature: it has been discovered that also oxadiazoles conjugating to sulfur atoms present interesting biological properties. Guogang et al. obtained promising results after the synthesis of a library of 1,2,4-triazoles and 1,3,4-oxadiazoles, including a series of 5-thioxo-1,3,4-oxadiazole analogs, differing for the kind of phenyl substituent and the type of thio-derivative introduced on the lateral chain in position 5 [29]. Comparing 1,2,4-triazoles and 1,3,4-oxadiazole analogs, 1,2,4-triazoles have lower toxicity. Indeed, the highest antiproliferative activity was recorded by
The presence of an aromatic system is not the only important structural feature: it has been discovered that also oxadiazoles conjugating to sulfur atoms present interesting biological properties. Guogang et al. obtained promising results after the synthesis of a library of 1,2,4-triazoles and 1,3,4-oxadiazoles, including a series of 5-thioxo-1,3,4-oxadiazole analogs, differing for the kind of phenyl substituent and the type of thio-derivative introduced on the lateral chain in position 5 [106]. Comparing 1,2,4-triazoles and 1,3,4-oxadiazole analogs, 1,2,4-triazoles have lower toxicity. Indeed, the highest antiproliferative activity was recorded by
65
(
Figure 5) on K562 cells, showing an 85% inhibition ratio [29].
12) on K562 cells, showing an 85% inhibition ratio [106].
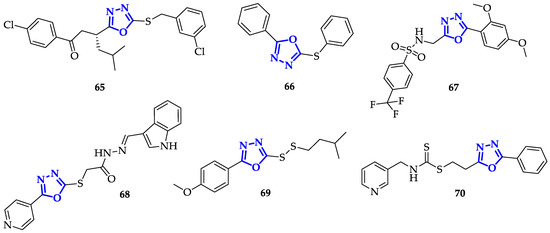
Figure 512.
In an attempt to improve both bioavailability and the antitumor effects, Kumar et al. synthesized different 2,5-disubstituted-1,3,4-oxadiazoles, containing both sulfonamide moiety and fluorine atoms, to increase lipophilicity and cellular uptake [30]. Sulfonamide groups, present in several FDA-approved drugs, were often used to improve drug-like design [31][32]. In this context,
In an attempt to improve both bioavailability and the antitumor effects, Kumar et al. synthesized different 2,5-disubstituted-1,3,4-oxadiazoles, containing both sulfonamide moiety and fluorine atoms, to increase lipophilicity and cellular uptake [108]. Sulfonamide groups, present in several FDA-approved drugs, were often used to improve drug-like design [86,109]. In this context,
67
(
Figure 5) stood out for a drastic reduction in cell survival percentage, which was below 30% in the case of K562 cells and 42.52% and 40.68% with Colo-205 and MDA-MB 231, respectively. This result suggests that the presence of electron-donating substituents on this kind of scaffold increased anti-cancer toxicity.
12) stood out for a drastic reduction in cell survival percentage, which was below 30% in the case of K562 cells and 42.52% and 40.68% with Colo-205 and MDA-MB 231, respectively. This result suggests that the presence of electron-donating substituents on this kind of scaffold increased anti-cancer toxicity.
Another interesting functional group involving sulfur atom is represented by dithiocarbamate, recently emerged for its promising anticancer properties [33]. With the aim to improve anticancer efficacy, Li et al. synthesized novel heteroarylmethylcarbamodithioates containing 1,3,4-oxadiazole scaffold, and evaluated their biological activity on hepatoma cells Bel-7402, breast cancer cells SK-BR-3 and MDA-MB-468 [34]. During this screening, compound
Another interesting functional group involving sulfur atom is represented by dithiocarbamate, recently emerged for its promising anticancer properties [112]. With the aim to improve anticancer efficacy, Li et al. synthesized novel heteroarylmethylcarbamodithioates containing 1,3,4-oxadiazole scaffold, and evaluated their biological activity on hepatoma cells Bel-7402, breast cancer cells SK-BR-3 and MDA-MB-468 [113]. During this screening, compound
70
(
Figure 5) resulted to be the most toxic with marked preference for Bel-7402 (IC
12) resulted to be the most toxic with marked preference for Bel-7402 (IC
50
= 1.23 ± 0.49 µM) and SK-BR-3 (IC
50
= 0.58 ± 0.05 µM) cells. Due to its promising activity,
70
has been further tested against other cancer cell lines, including lung cancer (H1299, H460, H522), hepatoma (Bel-7402), breast (MDA-MB-468, SK-BR-3), colorectal (COLO205, HCT-8, HCT-116, SW620) and prostate (DU145, PC3) in comparison with non-malignant cell lines (LO2 and 293).
70
presented good toxicity in all cancerous cells, with IC
50
values from 0.39 to 7.91 µM, while IC
50
values above 9 µM were measured in normal cells. Moreover,
70 caused chromatin condensation, a clear sign of induced apoptosis, and stopped cell cycle at G2/M phase, both in Bel-7402 and PC-3 cells. Further analysis evidenced that this substrate has a mechanism of action similar to the well-known Taxol [35]; indeed, treatment with
caused chromatin condensation, a clear sign of induced apoptosis, and stopped cell cycle at G2/M phase, both in Bel-7402 and PC-3 cells. Further analysis evidenced that this substrate has a mechanism of action similar to the well-known Taxol [114]; indeed, treatment with
70
increased levels of MPM-2 and phosphorylated histone H3, two specific mitotic molecular markers that indicate mitotic arrest. This was further confirmed by the generation of Supernumerary Centrosomes and Multipolar Spindles formation upon treatment with
70
, an abnormal behavior observed also with Taxol, highlighting
70 as a promising candidate for the treatment of different kinds of tumors [34].
as a promising candidate for the treatment of different kinds of tumors [113].
As an alternative to previously reported approaches, modification with carbohydrates is a common strategy used to improve the activity of bioactive molecules and it has been exploited also in the case of 1,2,4-oxadiazoles. Avanzo et al. reported some 1,2,4-oxadiazoles conjugated to D-ribofuranoside [36] and tested their antiproliferative activity against A549, HBL-100, HeLa, SW1573, T-47D, and WiDr cancer cell lines. However, also these compounds presented only a moderate anticancer effect, with IC
As an alternative to previously reported approaches, modification with carbohydrates is a common strategy used to improve the activity of bioactive molecules and it has been exploited also in the case of 1,2,4-oxadiazoles. Avanzo et al. reported some 1,2,4-oxadiazoles conjugated to D-ribofuranoside [115] and tested their antiproliferative activity against A549, HBL-100, HeLa, SW1573, T-47D, and WiDr cancer cell lines. However, also these compounds presented only a moderate anticancer effect, with IC
50
ranging from 4 µM to over 100 µM.
Another interesting functionalization of 1,3,4-oxadiazoles scaffold is the conjugation of fatty acid analogs [37]. Hassan and co-workers described the synthesis of a library of 1,3,4-oxadiazol-2(3H)-one with fatty acids with different chain-lengths and numbers of double bonds, chosen for their promising anticancer properties [38]. Antiproliferative activity of each molecule has been tested against HeLa, MDA-MB-231, and KCL-22 (Lymphoblastoid) cells for 48 h. Authors measured IC
Another interesting functionalization of 1,3,4-oxadiazoles scaffold is the conjugation of fatty acid analogs [116]. Hassan and co-workers described the synthesis of a library of 1,3,4-oxadiazol-2(3H)-one with fatty acids with different chain-lengths and numbers of double bonds, chosen for their promising anticancer properties [117]. Antiproliferative activity of each molecule has been tested against HeLa, MDA-MB-231, and KCL-22 (Lymphoblastoid) cells for 48 h. Authors measured IC
50
values in the 6–50 µM range, with the highest toxicity observed in presence of
71
(IC
50
= 6.3 ± 1.1 µM on HeLa cells; IC
50
= 8.3 ± 1.2 µM on KCL-22; IC
50
= 9.6 ± 1.2 µM on MDA-MB-231;
Figure 6), characterized by a 16-carbon atoms chain covalently bound to 1,3,4-oxadiazol-2(3H)-one. This result underlined that the presence of a cis-double bond and a hydroxy group are two key structural features to guarantee more potent activity, despite the toxicity of
13), characterized by a 16-carbon atoms chain covalently bound to 1,3,4-oxadiazol-2(3H)-one. This result underlined that the presence of a cis-double bond and a hydroxy group are two key structural features to guarantee more potent activity, despite the toxicity of
71
resulted to be lower compared to reference compound doxorubicin. Moreover, fluorescence microscopy micrographs, in agreement with previous experiments, showed that
71
was able to induce apoptosis with higher efficiency compared to other compounds of the library.
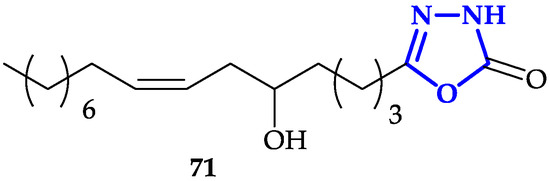
Figure 613.
In this context, several signs of progress were accomplished in the past years as confirmed by positive results described above. However, to develop novel and specific drug-like, a lot of effort is still required to identify other modifications and structural elements that could guarantee not only high antitumor potency but also interaction with specific and defined targets.