Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Takahiro Matsuyama.
Asthma is a heterogeneous disease characterized by chronic airway inflammation. Group 2 innate lymphoid cells (ILC2) play an important role in the pathogenesis of asthma. ILC2s lack antigen-specific receptors and respond to epithelial-derived cytokines, leading to the induction of airway eosinophilic inflammation in an antigen-independent manner. Additionally, ILC2s might be involved in the mechanism of steroid resistance. Numerous studies in both mice and humans have shown that ILC2s induce airway inflammation through inflammatory signals, including cytokines and other mediators derived from immune or non-immune cells.
- airway inflammation
- asthma
- comorbidity
1. Introduction
Asthma is a heterogeneous disease, characterized by chronic airway inflammation, variable expiratory airflow limitation, and a history of respiratory symptoms, such as wheezing, shortness of breath, chest tightness, and cough, which vary in intensity and over time [1]. It affects approximately 300 million people worldwide, with the prevalence increasing in developed countries.
Recent clinical and translational research has demonstrated that asthma is a heterogeneous disease comprising various phenotypes and endotypes. In terms of phenotype, asthma encompasses eosinophilic asthma, neutrophilic asthma, mixed granulocytic asthma, and pauci-granulocytic asthma [2]. Mixed granulocytic asthma is a phenotype characterized by increased levels of both eosinophils and neutrophils. Pauci-granulocytic asthma is a phenotype characterized by normal levels of both eosinophils and neutrophils [3]. Among these phenotypes of asthma, eosinophilic asthma, characterized by eosinophilia in the airways or blood driven by type 2 immune responses, can be induced by allergic and non-allergic mechanisms, mainly involving T helper type 2 (Th2) cells and group 2 innate lymphoid cells (ILC2), respectively.
Over the past decade, ILCs have been identified as a component of the innate immune system that can interact with various hematopoietic and non-hematopoietic cells to coordinate immunity, inflammation, and homeostasis in multiple organs throughout the body [4]. Unlike T cells and B cells, ILCs lack antigen-specific receptors and lineage (Lin) markers, and they cause antigen-non-specific immune responses [5,6][5][6]. ILCs consist of three subsets—ILC1s, ILC2s, and ILC3s—which are characterized by their transcription factors and the cytokines that they produce. These subsets correspond to Th1, Th2, and Th17 cells, respectively. Among these ILCs, ILC2s have been discovered in the gut, spleen, liver, and bone marrow [5,7,8][5][7][8]. ILC2s produce a large amount of IL-5 and IL-13 in response to epithelial-cell-derived cytokines, such as IL-25, IL-33, and thymic stromal lymphopoietin (TSLP), which are considered to play an essential role in the pathogenesis of allergic disorders, including asthma.
Currently, to achieve asthma control and to reduce the risk of exacerbations, various types of biologics have been used. Among biologics, dupilumab and tezepelumab target IL-4Rα and TSLP, respectively, and IL-4 and TSLP acted on ILC2s directly in in vivo or in vitro studies; therefore, these biologics might attenuate ILC2-mediated asthma. In a study using sorted peripheral blood ILC2s in vitro, the expression of type 2 cytokine mRNA was shown to be significantly decreased in asthmatics treated with dupilumab [119][9].
2. Airway Inflammation in Asthma Pathogenesis
The characteristics of asthma are chronic airway inflammation and airway hyperresponsiveness (AHR). Airway inflammation involves various inflammatory cells, such as eosinophils, neutrophils, lymphocytes, and mast cells, as well as airway structural cells including airway epithelial cells, fibroblasts, and airway smooth muscle cells, along with various humoral factors [9][10]. Persistent airway inflammation causes airway remodeling, which leads to irreversible airflow limitations. Furthermore, AHR is considered to be primarily induced by airway inflammation, whereas AHR is induced even when airway inflammation is mild with airway remodeling, because repeated bronchoconstriction promotes airway remodeling [10][11]. The airways of asthmatic patients exhibit pathologies, such as goblet cell metaplasia, excessive subepithelial collagen deposition, airway smooth muscle hyperplasia, and increased vascularity [11][12]. These findings are considered to be caused by inflammatory mediators, including cytokines and chemokines, produced by inflammatory cells and airway structural cells (Figure 1).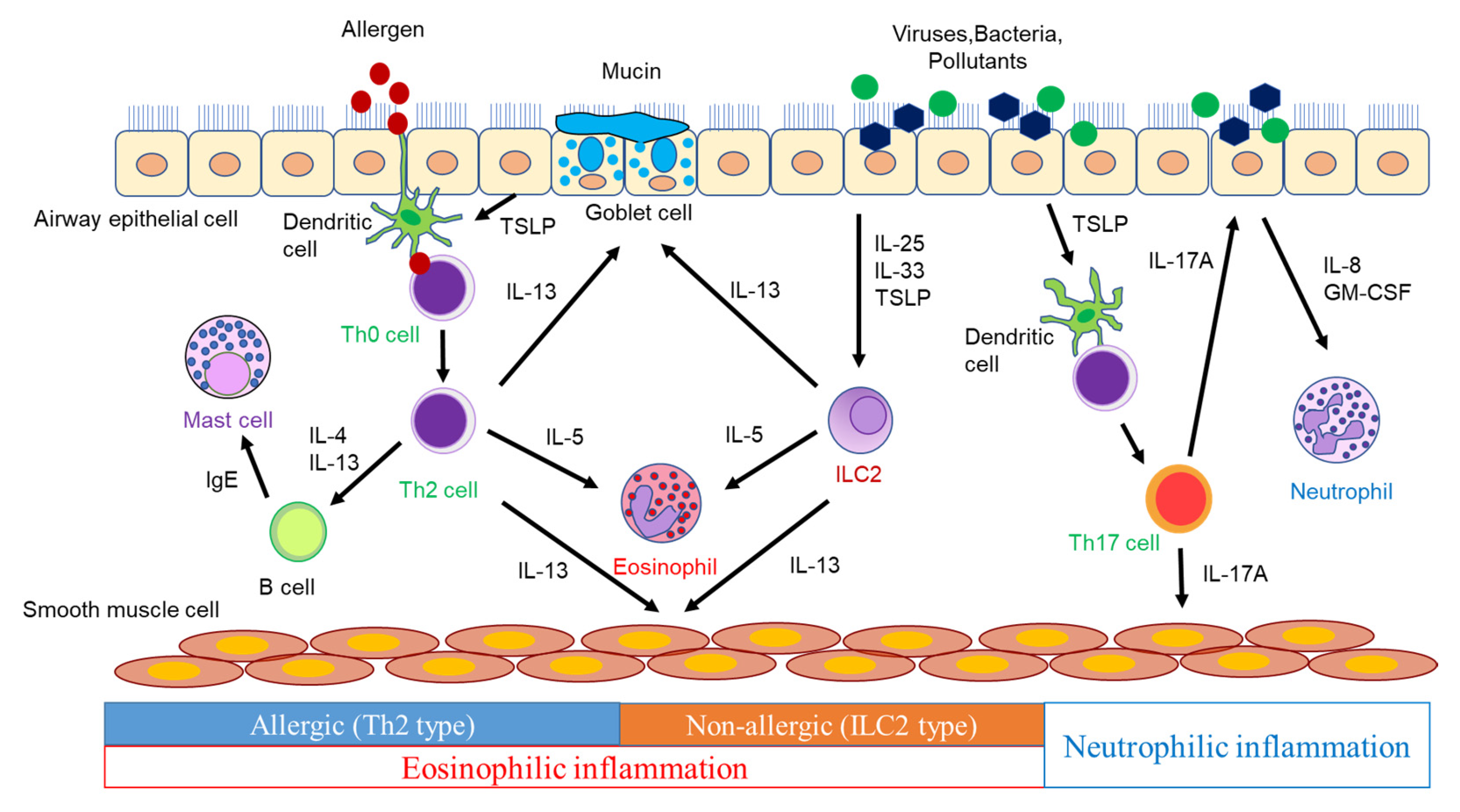
Figure 1.
The pathogenesis of airway inflammation in asthma.
References
- Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention 2022; GINA: Bethesda, MD, USA, 2022; Available online: http://ginasthma.org (accessed on 23 November 2022).
- Brusselle, G.; Bracke, K. Targeting immune pathways for therapy in asthma and chronic obstructive pulmonary disease. Ann. Am. Thorac. Soc. 2014, 11, S322–S328.
- Porsbjerg, C.; Lund, T.K.; Pedersen, L.; Backer, V. Inflammatory subtypes in asthma are related to airway hyperresponsiveness to mannitol and exhaled NO. J. Asthma 2009, 46, 606–612.
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293.
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate production of TH2 cytokines by adipose tissue-associated c-Kit+Sca-1+ lymphoid cells. Nature 2010, 463, 540–544.
- Kabata, H.; Moro, K.; Koyasu, S.; Asano, K. Group 2 innate lymphoid cells and asthma. Allergol. Int. Off. J. Jpn. Soc. Allergol. 2015, 64, 227–234.
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370.
- Price, A.E.; Liang, H.E.; Sullivan, B.M.; Reinhardt, R.L.; Eisley, C.J.; Erle, D.J.; Locksley, R.M. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 11489–11494.
- Patel, G.; Pan, J.; Ye, L.; Shen, X.; Rosloff, D.; D’Souza, S.S.; Fung, I.T.H.; Celstin, J.; Sun, W.; Sankar, P.; et al. Blockade of IL-4Rα inhibits group 2 innate lymphoid cell responses in asthma patients. Clin. Exp. Allergy 2020, 50, 267–270.
- Yasuda, Y.; Nagano, T.; Kobayashi, K.; Nishimura, Y. Group 2 Innate Lymphoid Cells and the House Dust Mite-Induced Asthma Mouse Model. Cells 2020, 9, 1178.
- Grainge, C.L.; Lau, L.C.; Ward, J.A.; Dulay, V.; Lahiff, G.; Wilson, S.; Holgate, S.; Davies, D.E.; Howarth, P.H. Effect of bronchoconstriction on airway remodeling in asthma. N. Engl. J. Med. 2011, 364, 2006–2015.
- Lambrecht, B.N.; Hammad, H.; Fahy, J.V. The Cytokines of Asthma. Immunity 2019, 50, 975–991.
- Maes, T.; Joos, G.F.; Brusselle, G.G. Targeting interleukin-4 in asthma: Lost in translation? Am. J. Respir. Cell Mol. Biol. 2012, 47, 261–270.
- Brusselle, G.G.; Koppelman, G.H. Biologic Therapies for Severe Asthma. N. Engl. J. Med. 2022, 386, 157–171.
- Chiba, Y.; Nakazawa, S.; Todoroki, M.; Shinozaki, K.; Sakai, H.; Misawa, M. Interleukin-13 augments bronchial smooth muscle contractility with an up-regulation of RhoA protein. Am. J. Respir. Cell Mol. Biol. 2009, 40, 159–167.
- Peters, M.C.; Wenzel, S.E. Intersection of biology and therapeutics: Type 2 targeted therapeutics for adult asthma. Lancet 2020, 395, 371–383.
- Yang, L.; Anderson, D.E.; Baecher-Allan, C.; Hastings, W.D.; Bettelli, E.; Oukka, M.; Kuchroo, V.K.; Hafler, D.A. IL-21 and TGF-beta are required for differentiation of human TH17 cells. Nature 2008, 454, 350–352.
- McKinley, L.; Alcorn, J.F.; Peterson, A.; Dupont, R.B.; Kapadia, S.; Logar, A.; Henry, A.; Irvin, C.G.; Piganelli, J.D.; Ray, A.; et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immunol. 2008, 181, 4089–4097.
- Hynes, G.M.; Hinks, T.S.C. The role of interleukin-17 in asthma: A protective response? ERJ Open Res. 2020, 6, 00364–02019.
- Busse, W.W.; Holgate, S.; Kerwin, E.; Chon, Y.; Feng, J.; Lin, J.; Lin, S.L. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am. J. Respir. Crit. Care Med. 2013, 188, 1294–1302.
- Halim, T.Y.; Krauss, R.H.; Sun, A.C.; Takei, F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity 2012, 36, 451–463.
- Hsu, A.T.; Gottschalk, T.A.; Tsantikos, E.; Hibbs, M.L. The Role of Innate Lymphoid Cells in Chronic Respiratory Diseases. Front. Immunol. 2021, 12, 733324.
More