Relevant, predictive normal, or disease model systems are of vital importance for drug development. The difference between nonhuman models and humans could contribute to clinical trial failures despite ideal nonhuman results. As a potential substitute for animal models, human induced pluripotent stem cell (hiPSC)-derived cardiomyocytes (CMs) provide a powerful tool for drug toxicity screening, modeling cardiovascular diseases, and drug discovery.
- hiPSC-derived cardiomyocytes
- maturation
- subtype
- disease modeling
1. Introduction
Human induced pluripotent stem cells (hiPSCs) were developed by Dr. Shinya Yamanaka more than 10 years ago [1]. This technology allows pluripotent stem cells to be derived from healthy persons, as well as patients. hiPSCs have been used in multiple fields, leading to significant technological and therapeutic developments. hiPSC-derived cardiomyocytes (CMs) have been used to model several major cardiomyopathies, including ion related, structural, and metabolic cardiomyopathy, providing new insights into the mechanism underlying the disease phenotype. A potential genetic therapy based on CRISPR/Cas9 and adeno-associated virus has also been proposed and validated in an hiPSC disease model. Another promising application of hiPSC-CMs is drug toxicity screening (Figure 1); despite the remaining issues such as immaturity and heterogeneity within the hiPSC-derived CM culture, a new paradigm based on hiPSC-CMs has been proposed for more accurate prediction of the proarrhythmia risk.
In this review, we provide an overview of hiPSC-CMs and their features, including characterization, maturation, and tissue engineering. Their applications in cardiac disease modeling and new drug testing paradigms are also summarized and discussed.
2. Generation of Human iPSC-CMs and Their Subtypes
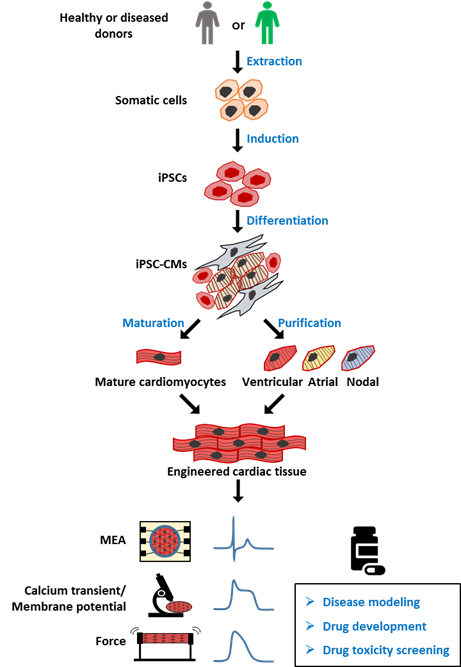
Figure 1. Overview of human induced pluripotent stem cell-derived cardiomyocyte (hiPSC-CM) model for drug screening. Healthy or patient-derived somatic cells can be reprogrammed into human induced pluripotent stem cells (hiPSCs) and then differentiated into all subtypes of cardiomyocytes (hiPSC-CMs), including ventricular, atrial, and nodal myocytes. iPSC-CMs can be matured and engineered into three-dimensional (3D) cardiac tissue, and used for applications including disease modeling, drug development, and toxicity screening. MEA: multielectrode array, for detection of extracellular field potential (FP) of CMs. Calcium transient: the intracellular calcium concentration during the CMs beating. Membrane potential: difference in electric potential between the inside and outside of cell membrane. The recording of membrane potential of CMs can be used for analyzing action potential (AP). Force: also known as contractile force, generated by the shift of the sarcomere. The contractile force and frequency are closely related to cell function.
Cardiovascular disease (CVD) is a leading cause of the global deaths [2]. Modeling CVD is essential for understanding its causes and the therapies of such diseases. There are already reports on the use of human primary CMs to model human heart [3][4][5][6][3–6]; however, limited access to human samples and the variability of human material cause problems, since each tissue source can only be assessed once [3]. hiPSC-CMs can be obtained in large amounts, and they recapitulate the properties of human heart cells. Ventricular CMs, a chamber-specific CM population, have been differentiated with high purity and widely used in the study of drug responses and disease modeling. Notably, a chemically defined cardiac differentiation protocol was recently developed to produce ventricular-like CMs with >90% purity and on a large scale [7]. The heart is composed of multiple cell subtypes, including not only ventricular CMs, but also pacemaker cells and atrial myocytes [8]. These subtypes are all important to the proper functioning of heart. In order to obtain more accurate drug responses and better therapeutic effects, it is of vital importance to acquire tissue-specific cells and promote their maturation [9][10][9,10]. While significant progress has been achieved in ventricular tissue engineering, iPSC-derived atrial tissues are still immature.
In contrast to ventricular cardiomyocytes, atrial cells are smaller and thinner, and they have fewer transverse tubules (T-tubules) and less calcium-handling machinery. Retinoic acid (RA) was recently used to differentiate iPSCs into atrial CMs. Lee et al. developed an improved differentiation protocol for the generation of atrial linages by utilizing developmental signaling gradients that specify atrial mesoderm precursors [11]. More recently, by using a stage-specific activation of RA signaling in monolayer-based culture, Cyganek et al. demonstrated that cardiac progenitors could be efficiently directed toward a highly homogeneous population of atrial CMs [12]. Zhao et al. described a scalable tissue-cultivation platform that can electrophysiologically distinguish atrial and ventricular tissues with chamber-specific drug responses and gene expression [13]. These studies provide a solid foundation for further generation of atrial tissues.
Pacemakers are important for patients suffering from cardiac arrhythmia. There are numerous reports on using different protocols to obtain and evaluate cell populations from the cardiac conduction system. White et al. successfully developed a cardiac pacemaking conduction system via co-expression of the chicken GATA6 enhancer and mink-lacZ transgene [14]. Yano et al. also reported that a fraction of Nkx2.5-positive cardiac precursor cells were committed to pacemaker cells expressing If channels predominantly encoded by the hyperpolarization-activated cyclic nucleotide-gated 1 (HCN1) and HCN4 genes [15]. It was reported that the addition of B12 or SKCa activator during differentiation can promote an increase in the nodal population [16]. Moreover, via stage-specific manipulation of developmental signaling pathways, Protze et al. developed a transgene-independent protocol to differentiate sinoatrial node cells from iPSCs [17]. Further efforts are needed to develop a robust differentiation protocol for generating mature ventricular CMs, atrial CMs, and pacemakers to enable better myocardium recapitulation.
3. Patient-Specific iPSC-CMs as Disease Models
iPSCs have been derived from patients and introduced into various patient-specific iPSC-CMs for modeling cardiomyopathies in vitro. These models can be categorized as inherited or nonhereditary, and they are reviewed below (Table 1).
3.1. Inherited Cardiomyopathy
3.1.1. Ion Channelopathy
Ion channelopathies are one of the most well-established iPSC-based disease models because of their better-understood impact on action potential (AP) [18]. Abnormalities occurring in AP generation, synchronization, or propagation may cause cardiac channelopathies related to arrhythmia [19]. The most common ion channelopathy is long QT syndrome (LQTS), which has a prevalence of 1 in 2000 [20]. Decreased systolic Ca2+ release leads to impaired cellular contractility and delayed repolarization of ventricular CMs. LQTS is characterized by a prolonged QT interval, causing increased risk of ventricular tachyarrhythmia or sudden cardiac death [21]. LQTS is divided into more than 10 different subtypes defined by specific ion channel mutations. Among these, the voltage-gated sodium (Nav) channel and the cardiac voltage-gated potassium (Kv) channel, both electrical impulse-initiating ion channels, are the primary mutation types [22]. LQTS type 1 (LQT1) occurs due to mutations in KCNQ1, and LQT1 iPSC-CM disease modeling was first generated by Moretti and his collages [23]. Different disease-specific human iPSC lines have been developed from patients with these ion channel gene mutations. The LQT1 disease model accurately reflects the disease features, having a slow outward potassium current (IKs), abnormal channel activities, and increased susceptibility to tachyarrhythmia induced by catecholamine [24][25][24,25]. LQT2 has mutations in KCNH2, a human ether-à-go-go-related gene (hERG) that mediates rapid delayed-rectifier potassium current IKr, which is important for the repolarization phase of the AP [26]. Thus, the LQT2 disease models revealed significant prolongation of the action potential duration (APD) and a reduction in IKr when compared to healthy control cells [27]. Precise genetic modification of the KCNH2 mutation increased the IKr current conducted by the hERG channel and normalized the APD [28]. Malan et al. developed hiPSC-CMs from an LQT3 patient with an SCN5A mutation, known to mediate fast Nav1.5 channel inactivation. LQT3 hiPSC-CMs exhibited accelerated recovery from Nav1.5 inactivation, AP prolongation, and early afterdepolarizations (EADs) even at low stimulation rates, which are considered to be the main cause of arrhythmia [29]. Roche et al. investigated the SCN5A mutation, using different independent systems, and compared the advantages and limitations of disease-specific, engineered iPSC-CMs and heterologous HEK293-cells for disease modeling and drug discovery, emphasizing the importance of investigating the mechanisms of Brugada syndrome in independent systems [30]. These cell-based models indicated that ion-trafficking defects are the associated pathological mechanism of the disease electrophysiological phenotype, and that regulation of key genes may be governed by a complex regulatory landscape.
Another common inherited channelopathy is catecholaminergic polymorphic ventricular tachycardia (CPVT), mainly caused by mutations in calcium-handling genes characterized by Ca2+ cycling and electrophysiology defects in patients [31]. The most prevalent CPVT1, responsible for 60% of total cases, is caused by mutations in RYR2, which encodes the cardiac ryanodine receptor. CPVT2 is less common, causing less than 5% of total cases, and is produced by a mutation in CASQ2, encoding cardiac calsequestrin [32]. Both mutations lead to abnormal calcium leakage from the sarcoplasmic reticulum (SR), causing cytosolic calcium overload and subsequent delayed afterdepolarizations and triggering ventricular arrhythmias [33]. CPVT patient-derived hiPSC-CMs carrying either RYR2 or CASQ2 mutations have been generated by several groups. Using selective pharmacology and genome editing, Park et al. generated a novel model that effectively recapitulates the CPVT1 profile caused by dominant mutations in RYR2 [34]. They regarded the activation of Ca2+/calmodulin-dependent protein kinase (CaMK II) as a key factor for triggering arrhythmias in CPVT patients, suggesting a molecular pathway linking β-adrenergic stimulation to arrhythmogenesis. Using these disease models, a series of potential compounds have been tested for modifying aberrant Ca2+ handling and delayed afterdepolarizations (DADs) [35][36][37][38][35–38].
3.1.2. Structural Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is a common inherited heart disease with abnormalities in morphology, with an estimated prevalence of 1 in 500 worldwide [39]. Most cases of sudden death related to HCM are caused by the conversion of ventricular arrhythmia to ventricular fibrillation [40]. Over 1500 mutations have been identified in HCM, most of which are located in sarcomere genes. These genes are responsible for CM contraction and relaxation [41]. Approximately 70% of HCM patients had either MYH7 (encoding β-myosin heavy chain) or MYBPC3 (myosin-binding protein) mutations, and their heart explants revealed lower tension forces compared to healthy individuals [42]. Less common mutations are located in other sarcomere genes such as actin (ACTC), cardiac troponin T (TNNT2), myosin light chain (MYL2), and cardiac troponin I (TNNI3) or non-sarcomere genes such as ion channels, Z-disc genes, and membrane transporters [41]. Five HCM hiPSC-CM models have been derived from patients carrying either MHY7 or MYBPC3 mutations using viral vectors [43]. Intraventricular injection of adeno-associated virus has shown potential as therapy for treating the MYL2 mutation in heart cells [44]. CRISPR/Cas9 editing has also been used to generate HCM disease modeling with site-directed homozygous or heterozygous variants [45]. These cell-based models recapitulate key features of the HCM phenotype such as increased sarcomere organization and aberrant Ca2+ handling, providing a new in vitro model for identifying pathogenesis and developing new therapeutic strategies for these inherited heart disease [46]. However, engineered heart tissue (EHT) has more advantages in terms of mechanism elucidation than single-cell models, as EHT can better reflect and mimic the cell–cell interaction at the tissue level. Cashman et al. first developed EHTs created from cardio-facio-cutaneous syndrome (CFCS) patients with BRAF mutations (encoding a serine/threonine kinase), and this tissue-based model better recapitulated key aspects of the HCM phenotype in vivo, providing a powerful tool for studying the patient-specific mechanisms of myocardial dysfunction [47].
Dilated cardiomyopathy (DCM) is another type of structural cardiomyopathy. DCM is mainly due to sarcomere mutations and has a prevalence of 1 in 2500 individuals [48]. Hearts affected by DCM tend to have increased chamber size and thinner chamber walls leading to volume overload, systolic dysfunction, and progressive heart failure (HF) [49]. DCM has high morbidity and mortality rates and is the leading cause of HF in young people. More than 80 different genes associated with DCM have been described. Of these, TTN encoding titin is the most prevalent mutant gene identified in around 20–25% of DCM patients [50]. Sun et al. first developed DCM iPSC-CMs with a mutation in TNNT2, recapitulating the DCM disease phenotypes morphologically and functionally. Their model has altered Ca2+ handling, decreased contractility, and abnormal α-actin distribution [51]. Dai et al. revealed that the TNNT mutation destabilizes the molecular interactions of troponin with tropomyosin and limits PKA binding to sarcomere [52]. Mutations occur less commonly in nuclear lamina, Nav channel α-subunit 5 (SCN5A), desmin (DES), phospholamban (PLN), Bcl2-associated athanogene 3 (BAG3), and RNA-binding motif protein 20 [53].
Arrhythmogenic cardiomyopathy (ACM) is another common structural cardiomyopathy usually caused by mutations in desmosomal proteins, leading to progressive HF and lethal arrhythmias [54]. Approximately half of the patients with ACM have gene mutations in desmosomes, such as desmoplakin (DSP), desmocollin (DSC), desmoglein-2 (DSG2), plakoglobin (JUP), and plakophilin-2 (PKP2). The PKP2 mutation is the most common pathogenic type in ACM [54]. hiPSC-CMs with a PKP2 mutation recapitulated key features of arrhythmogenic right-ventricular cardiomyopathy (ARVC), including low β-catenin activity, abnormal nuclear translocation of junction plakoglobin, and less cell surface localization of desmosomes, presenting an adipogenic phenotype [55]. However, it was reported by Kim et al. that only by co-activating peroxisome proliferator-activated receptor (PPAR)-α/PPAR-γ pathways, both of which are responsible for metabolism, can iPSC-CMs with a PKP2 mutation display efficient ACM features within 2 months [56][57][56,57]. This report proposed for the first time that induction of adult-like metabolism phenotype plays a role in adult-onset disease modeling.
Duchenne muscular dystrophy (DMD) is a rare X-linked recessive disease with an incidence of 1 per 5000 males. The cells of DMD patient are highly susceptible to mechanical stress and injury as they lack the dystrophin protein [58]. Dystrophin is a fundamental component of the dystrophin–glycoprotein complex, which is expressed at the muscle sarcolemma and bridges the cytoskeleton and extracellular matrix, maintaining cellular stability [59]. Dystrophin deficiency leads to progressive muscle scarring and degeneration, HF, and eventually death. DMD patient-derived iPSC-CMs exhibited excessive Ca2+ influx and increased sensitivity to hypotonic stress, accumulation of reactive oxygen species (ROS), and mitochondrial damage, eventually inducing cell apoptosis [60]. Dystrophin modifications by CRISPR/Cas9 have been proven to be a fast way to rescue DMD, with efficient restoration of CM contractility and calcium transients detected to varying degrees [61].
3.1.3. Metabolic Cardiomyopathy
Acid-α-glucosidase (GAA) is an amylolytic enzyme located in the lysosome and is responsible for glycogen degradation. Deficiency of GAA results in the accumulation of glycogen in lysosomes, a condition called Pompe disease (PD) [62]. As a result of dysregulation of glycogen metabolism, PD myocytes display increased cytoplasmic glycogen particles, endoplasmic reticulum stress, mitochondrial aberrance, abnormal calcium signaling, and progressive autophagic buildup [63]. The PD can be divided into infantile and late-onset phenotypes. Huang et al. reported that derivation of infantile-onset PD-iPSCs into CM-like cells recapitulated the hallmark of PD cells, including glycogen accumulation and differential ultrastructural aberrations [64]. A subsequent drug rescue test showed that GAA or I-carnitine could reverse the major pathologic phenotypes. Raval et al. investigated the mechanism of PD in tissue using a generated EHT model [65]. They stated that the lack of GAA ability leads to deficits in Golgi-based protein glycosylation, thus finally leading to lysosomal glycogen accumulation and HCM. Sato et al. generated late-onset Pompe disease-specific iPSC-CMs and showed that glycogen accumulation can be ameliorated by lentiviral GAA rescue [66]. Using metabolic profile analysis, they found that oxidative stress and mitochondrial dysfunction induced in the PD model may be related to cardiac complications [67]. The imbalance between oxidative stress and an antioxidative stress response may, therefore, reveal the pathogenesis of late-onset PD.
Barth syndrome (BTHS) is an X-linked mitochondrial disorder caused by a mutation in tafazzin, an acyltransferase encoded by TAZ [68]. Tafazzin is responsible for the normal acylation of cardiolipin, which is mainly located in the mitochondrial inner membrane. BTHS features multisystem disorders such as cardiomyopathy, neutropenia, and skeletal myopathy[69]. Using BTHS patient-derived iPSC-CMs, Wang et al. investigated the structural, metabolic, and functional abnormalities caused by TAZ mutation [70]. They engineered BTHS iPSC-CMs into a “heart-on-chip” and demonstrated sparse and irregular sarcomeres with weak contractile force in this chip. These findings indicate the presence of a link between TAZ mutation and impaired CM mechanical function, providing new insights into the pathogenesis of BTHS.
3.2. Chronic Nonhereditary Cardiomyopathy
Chronic heart failure (CHF), such as congestive heart failure, is a progressive syndrome caused by CVDs including coronary artery disease and myocardial infarction, as well as high blood pressure, and it results in structural or functional changes in the heart [71]. Heart failure with reduced ejection fraction (HFrEF) is a common type of CHF, usually caused by long-term use of catecholamines (e.g., norepinephrine) in patients with end-stage HF [72]. HFrEF hearts show hypertrophy, a weaker force–frequency response, and decreased β-adrenergic sensitization [73]. Through chronic norepinephrine stimulation, Tiburcy et al. successfully generated HF models with not only pathological hypertrophy, cellular death, and contractile dysfunction, but also N-terminal pro B-type natriuretic peptide (NT-proBNP) release, features consistent with the clinical diagnosis of HF [74]. Their work provides guidance for the establishment of HF modeling, drug screening, and tissue-based heart repair.
Table 1. Categories of patient-specific iPSC-CMs as disease models.
|
Disease Model Categories |
Related Genes |
Reference |
|||
|
Inherited cardiomyopathy |
Ion Channelopathy |
Long QT syndrome (LQTS) |
Type 1 |
KCNQ1 |
|
|
Type 2 |
KCNH2 |
||||
|
Type 3 |
SCN5A |
||||
|
Catecholaminergic polymorphic ventricular tachycardia (CPVT) |
RYR2 |
[34] |
|||
|
Structural Cardiomyopathy |
Hypertrophic cardiomyopathy (HCM) |
MYH7, MYBPC3, ACTC, TNNT2, MYL2, TNNI3 |
[43][43,45,47] |
||
|
Dilated cardiomyopathy (DCM) |
TTN, TNNT2, SCN5A, DES, PLN, BAG3 |
||||
|
Arrhythmogenic cardiomyopathy (ACM) |
DSP, DSC, DSG2 JUP, PKP2 |
[54][54,56] |
|||
|
Duchenne muscular dystrophy (DMD) |
DMD |
[60] |
|||
|
Metabolic cardiomyopathy |
Pompe disease (PD) |
GAA |
|||
|
Barth syndrome (BTHS) |
TAZ |
[70] |
|||
|
Chronic nonhereditary cardiomyopaty |
Chronic Heart Failure |
Heart failure with reduced ejection fraction (HFrEF) |
N/A |
[74] |
|