Nrf2 and its principal negative regulator, the E3 ligase adaptor Kelch-like ECH- associated protein 1 (Keap1), play a central role in the maintenance of intracellular redox homeostasis and regulation of inflammation. Interestingly, Nrf2 is proven to contribute to the regulation of the heme oxygenase-1 (HO-1) axis, which is a potent anti-inflammatory target. Recent studies showed a connection between the Nrf2/antioxidant response element (ARE) system and the expression of inflammatory mediators, NF-κB pathway and macrophage metabolism. Thus, Nrf2 is part of a multilayered network that plays a key role in redox homeostasis as well as in inflammation. This suggests a new strategy for designing chemical agents as modulators of Nrf2 dependent pathways to target the immune response.
- Nrf2
- inflammation
- Anti-Inflammatory Drugs
Dear author, the following contents are excerpts from your papers. They are editable.
(Due to the lack of relevant professional knowledge, our editors cannot complete a complete entry by summarizing your paper, so if you are interested in this work. you may need to write some contents by yourself. A good entry will better present your ideas, research and results to other scholars. Readers will also be able to access your paper directly through entries.)
1. Introduction
Nrf2 (NF-E2-related factor 2), a member of the Cap’n’collar (CNC) transcription factor family, consists of 605 amino acids and is divided into seven highly conserved functional domains, known as Neh1-Neh7 (Figure 1). The N-terminal domain influences the stability and ubiquitination of Nrf2 by its negative regulator Keap1, while the Neh5 domain is responsible for the cytoplasmic localization of Nrf2 [1][7]. The Neh1 domain has a cap ‘n’ collar basic-region leucine zipper (bZIP) domain, which regulates DNA-binding [2][8] and a nuclear localization signal (NLS) that is responsible for the nuclear translocation of Nrf2 [3][9]. The Neh3, Neh4, and Neh5 are transactivation domains mediating the interaction of Nrf2 with other coactivators [4][5][10,11]. Neh6 domain with serine-rich residues is a negative regulatory domain which binds to a β-transducin repeat-containing protein (β-TrCP) leading to Nrf2 ubiquitination [6][12]. The Neh7 domain inhibits Nrf2-ARE signaling pathway by promoting the binding of Nrf2 to the retinoic X receptor α (RXRα) [7][13].
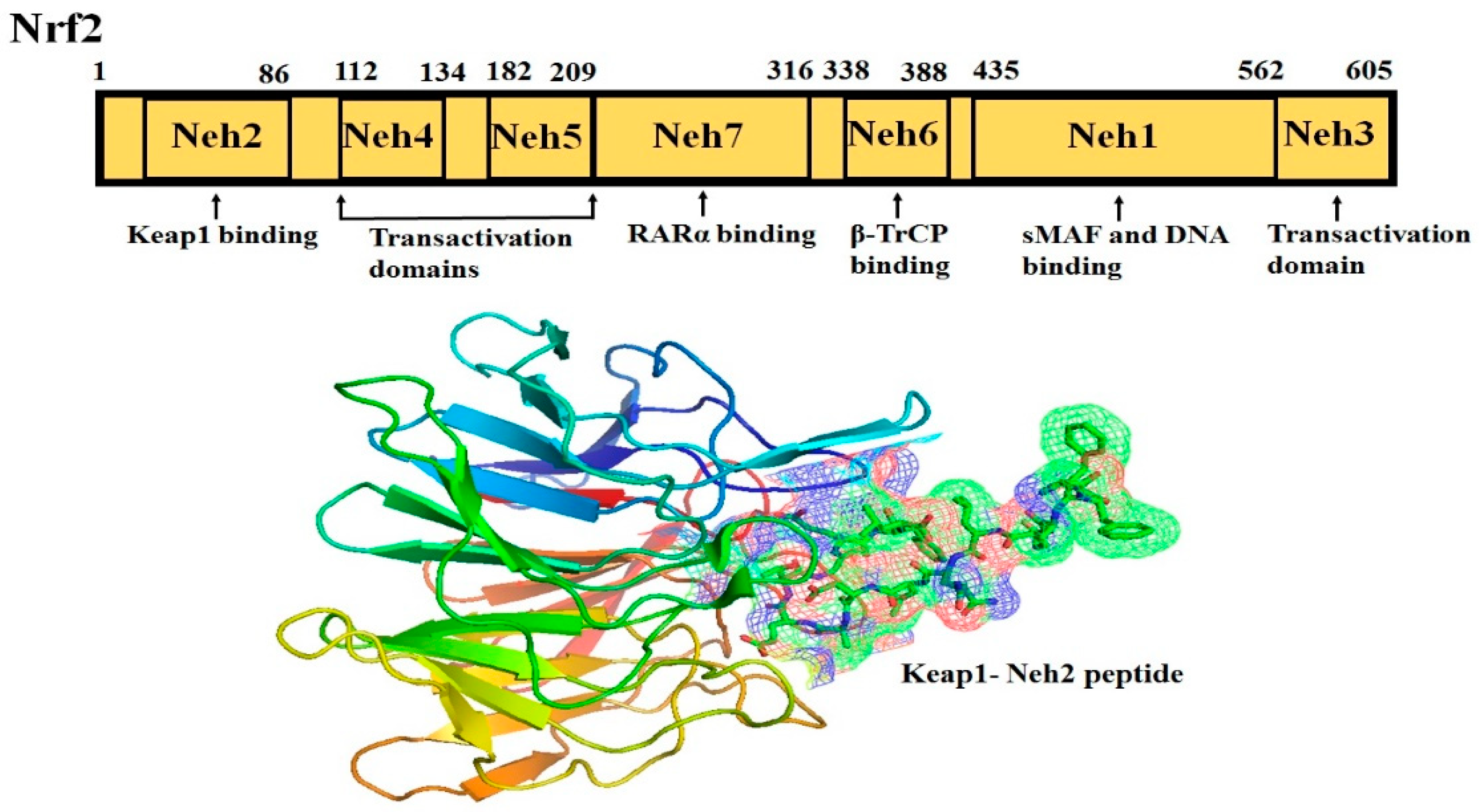
The Neh2 domain, an N-terminal regulatory domain, contains seven lysine residues that influence the ubiquitin conjugation [8][14] and two peptide binding motifs (ETGE and DLG) that regulate Nrf2 stability by promoting its binding to different proteins [9][15]. The ETGE and DLG motifs interact with Keap1, which is a substrate adaptor protein for the Cullin 3- dependent E3 ubiquitin ligase complex that facilitates Nrf2 ubiquitination and its proteasomal degradation under normal physiological conditions [9][10][11][15,16,17].
Keap1, consisting of 624 amino acids, is a cysteine-rich protein, containing 27 cysteine residues in humans [12][18]. Keap1 is divided into five domains, an N-terminal region (NTR), a Tramtrack and Bric-à-Brac (BTB) domain, a central intervening region (IVR) with a nuclear export signal (NES) mediating the cytoplasmic localization of Keap1 [13][19], six Kelch repeats, and a C-terminal domain (CTR) (Li et al., 2004). The BTB domain is responsible for Keap1 homodimerization and its binding to the cullin-based (Cul3) E3 ligase, leading to the formation of Keap1-Cul3-RBX1 (Ring box protein-1) E3 ligase complex [14][20], whereas Kelch repeats are thought to mediate the binding of Keap1 to Nrf2 and p62 (Figure 2) [15][16][21,22].
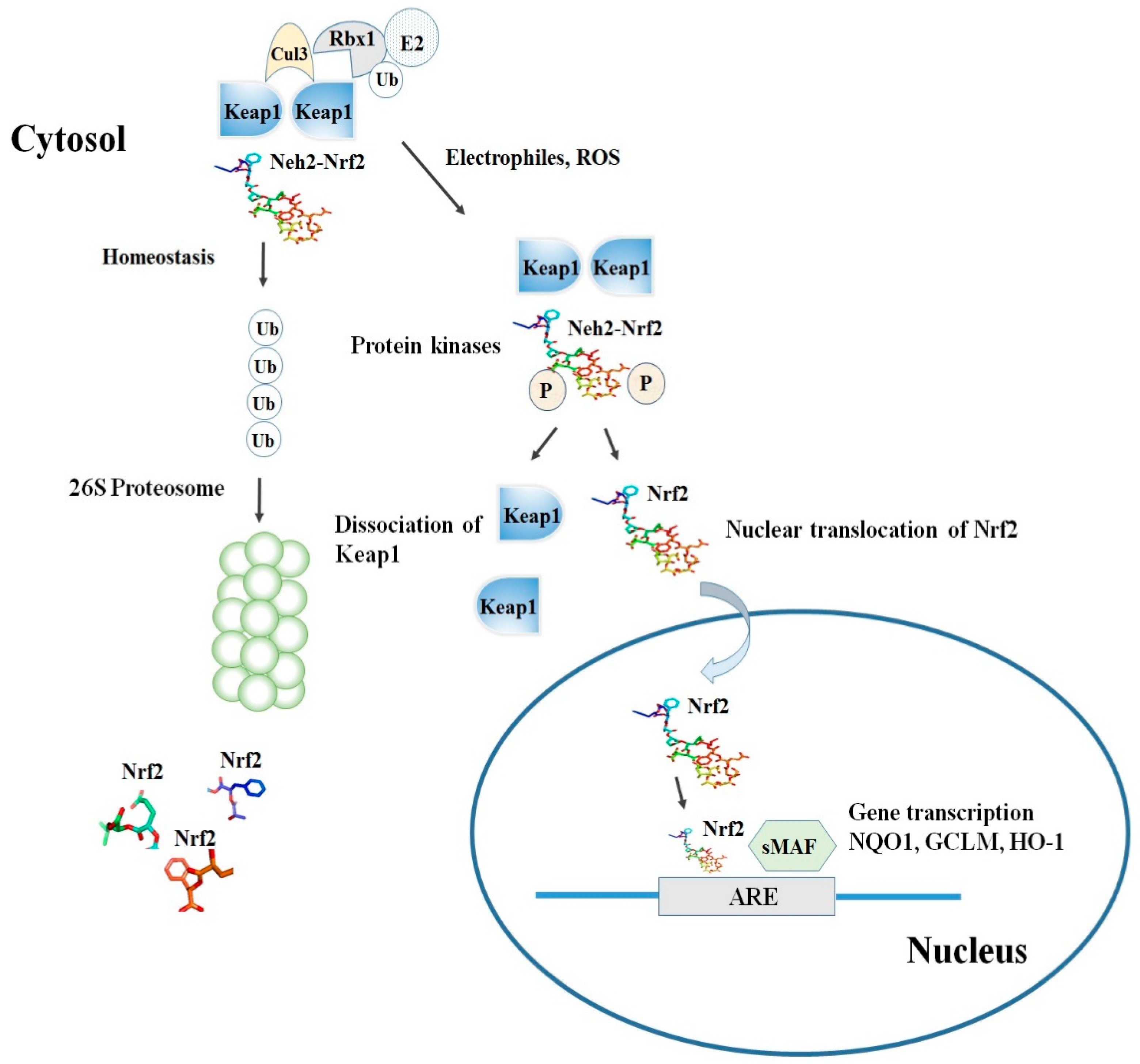
Among the others, the cysteine residues C151, C273, and C288 are highly reactive and susceptible of covalent modifications by ROS, RNS, H2S and other electrophiles. In this respect, the S-sulfenylation, the S-nitrosylation and the S-sulfhydration of these critical residues were shown to induce conformational changes of Keap1 that ultimately promote the dissociation of Nrf2 and its stabilization [17][18][19][23,24,25]. Although the exact mechanism of the Nrf2-Keap1 interaction is still unknown, two models were reported in the literature to explain the regulation of Nrf2 stability. The first, also known as the ‘‘hinge and latch’’ model, postulates that Keap1 interaction with the ETGE domain acts as a hinge while a weaker interaction with the DLG motif acts as a latch [20][26].
When specific thiol residues are modified by electrophiles, the DLG motif dissociates from Keap1 causing a disruption in the alignment of Nrf2 lysine residues that ultimately prevent its ubiquitination (Figure 2) [17][21][23,27]. Consequently, Nrf2 is released from Keap1-Cul3-RBX1 complex and translocates into the nucleus wherein it heterodimerizes with small Maf proteins (sMaf). After release, Nrf2 binds to EpRE in the presence of small Maf, and up-regulates electrophile response element (EpRE)-mediated transcription [22][28]. This further activates the transcription of a battery of genes containing an antioxidant response element (ARE) within their promoter region [21][27]. In addition, the carboxy-terminal domain of Neh3 can interact with the transcription coactivator, CHD6 (chromo-ATPase/helicase DNA-binding protein) [4][10] while Neh4 and Neh5 can interact with another transcriptional co-activator, CBP (cAMP-response- element-binding protein-binding protein) [5][11]. Finally, Neh4 and Neh5 can also bind to the nuclear cofactor RAC3/AIB1/SRC-3, further expanding the list of Nrf2 functional interactors, to promote the expression of Nrf2-targeted ARE genes [23][29]. Interestingly, as shown by several studies, thiol modifications of specifically C151 in the BTB domain might represent a stress sensing-mechanism that prevents the Keap1-Cul3 interactions and thus facilitates Nrf2 activation in response to adverse conditions [24][30].
The second model, also known as Keap1-independent regulation, postulates that in normal conditions, Neh6 domain binds to the DSGIS and DSAPGS motifs of the β-TrCP (Beta-transducin repeats-containing protein), which in turn is a substrate receptor for the Skp1-Cul1-Rbx1/Roc1 ubiquitin ligase complex that drivesNrf2 ubiquitination [25][31]. The phosphorylation of Nrf2 in the Neh6 domain by glycogen synthase kinase-3 regulates the recognition of Neh6 domain by β-TrCP [26][32].
2. Nrf2 Dependent Anti-Inflammatory Drugs
Nonsteroidal anti-inflammatory drugs (NSAIDs) including traditional non-selective NSAIDS (nsNSAIDs) and cyclo-oxygenase-2 selective NSAIDs (COXIBs), are one of the most common medication for inflammation [27][219]. The primary activity of NSAIDs are enacted by blocking prostaglandins (PGs) synthesis through the cyclooxygenase enzymes (COX-1 and COX-2) inhibition. Aspirin is the most commonly used NSAID with antioxidant activity and reportedly activate Nrf2-ARE pathway and HO-1 expression in primary human melanocytes [28][220] as well as in neuronal apoptosis [29][221]. Similarly, celecoxib up-regulates heme oxygenase-1 (HO-1) expression in macrophages and vascular smooth muscle cells via redox signaling [30][222]. Treatment of celecoxib demonstrated increases in H-ferritin and TrxR1 mRNA levels respectively in Nrf2 dependent mechanism [31][223]. A widely used NSAID, diclofenac also activates Nrf2 expression and downstream related genes [32][224]. In choroidal neovascularization, indomethacin or bromfenac induces translocation of Nrf2 into the nucleus and up-regulates HO-1 expression [33][225]. On the other hand, pantoprazole, a proton pump inhibitor (PPIs) protected from NSAIDs-induced injury by inducing activation of Nrf2 through inactivation of Keap1 and by increasing expression of HO-1 in human gastric epithelial and endothelial cells and in animal model of gastric injury [34][226].
In vitro and in vivo studies on NO-NSAID (nitric oxide-donating NSAID) hybrid drugs such as NCX-4016 (mNO-ASA, nitric oxide-donating aspirin) and the isomeric NCX-4040, suggest significant potential for activation of Nrf2 through nitrosylation of Keap1 [35][36][227,228]. Moreover, cellular bioactivation of pNO-ASA yields a quinone methide, therefore, an alternative mechanism of Nrf2 activation by mNO-ASA is via an electrophilic quinone [37][229]. Analogues of NCX-4016 that are activated to quinone methide and without NO group, also showed covalent modification of Keap1, and induced Nrf2 translocation to the nucleus [38][230]. Accumulating evidence also suggests that treatment with small molecules such as 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid- derivatives and sulforaphane induces Nrf2 activation and subsequently reduces significantly Th1 and Th17 cytokines including IL-6 by inhibiting recruitment of RNA polymerase II [39][231]. Similarly, pharmacological activation of mycophenolate mofetil, prevented the proinflammatory cytokines overexpression in a Nrf2 dependent mechanism [40][232]. In addition, short peptides such as casein glycomacropeptide hydrolysate were shown to have anti-inflammatory and antioxidant activity by increasing the Nrf2 nuclear translocation and HO-1 expression in RAW 264.7 macrophages and HepG2 cells [41][42][233,234]. Nrf2 induction by triazole derivatives inhibits pro- inflammatory cytokines release in different models of Huntington disease including primary mouse microglia, astrocytes, and Drosophila model and in cultured monocytes from human patients with huntington disease [43][44][235,236].
Several cell-derived metabolites such as itaconate and fumarate demonstrated anti-inflammatory responses in LPS- stimulated macrophages [45][237]. A cell-permeable derivative of itaconate, 4-octyl-itaconate (4-OI) demonstrated in vivo anti-inflammatory activity by induction of Nrf2 [45][237]. Here, the derivative of fumarate, dimethyl fumarate (DMF), is the only drug approved by US Food and Drug Administration and marketed by Biogen, as an anti-inflammatory therapeutic agent in multiple sclerosis with the ability to inhibit inflammation via Nrf2 antioxidant pathway [46][47][238,239]. In vivo studies in rodents had reported that DMF metabolite, monomethyl fumarate, activates Nrf2 by adduct formation at C151 in Keap1 in neuroinflammation [46][238]. Other commercial prodrugs of monomethyl fumarate such as diroximel fumarate is in a phase III clinical trial for multiple sclerosis and tepilamide fumarate, is in a phase II clinical trial for plaque psoriasis [48][240]. V ClinBio developed conjugate of monomethyl fumarate and eicosapentaenoic acid for simultaneous modulation of Nrf2 and NF-κB in cell lines and animal models of multiple sclerosis and psoriasis. Preclinical studies by Colby Pharmaceuticals developed a di- substituted hydroxylamine compound (OT551) which inhibits inflammation and oxidative stress by targeting Keap1 (OMEGA/NCT00485394). An ophthalmic solution of this compound protects from inflammation in retinal pigment epithelium and photoreceptors (OMEGA/NCT00485394).
3. Conclusions
Nrf2-Keap1 signaling pathway is the hallmark of redox signaling and controlled inflammation and plays an important role in cell metabolic flexibility. Here, we reviewed ongoing scientific literature about regulation of Nrf2 signaling pathway in different aspects of inflammation such as cytokines, chemokine releasing factors, MMPs, and other inflammatory mediators affecting the NF-kB and MAPK networks to control inflammation. However, there are still some mechanisms such as interactions between Nrf2 and JAK/STAT signaling that needs to be investigated. Although many pharmaceutical companies are currently targeting Keap1, the prime regulator of Nrf2, it is still challenging to enhance the targeting of these compounds and their activity against these multifactorial complex diseases. Natural Nrf2 activators derived from plant sources as well as synthetic anti-inflammatory drugs require further experimental validation. Research for efficient therapeutic agents promoting Nrf2 activation came up with some new drugs which have entered clinical trials and will undoubtedly provide advancement in the management of inflammation in the near future. However, efforts are still ongoing to find new small molecule Nrf2 inducers with the advantages of oral administration, high target specificity, safe, and high bioavailability.