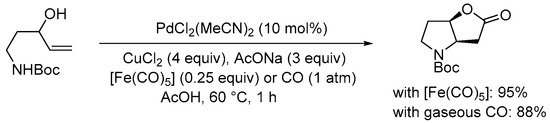Here, some examples of carbonylative double cyclization processes for the synthesis of carbonylated fused heterocycles (in particular, starting from suitably functionalized olefinic substrates) are presented.
2. Functionalized Olefinic Substrates
It is well-know that palladium(II)-based catalysts activate unsaturated carbon–carbon bonds towards the attack of a variety of nucleophilic groups (mainly oxygen- or nitrogen-based). The intramolecular version of this reactivity is of particular importance as it allows the construction of heterocyclic derivatives in a straightforward manner and under mild reaction conditions (Pd(II)-catalyzed heterocyclization reactions)
[3][5]. On the other hand, it is also very well known that Pd(II) catalysts promote many important kinds of carbonylation processes, particularly under oxidative conditions, including cyclization processes in which carbon monoxide is inserted as a carbonyl function inside the newly formed ring (cyclocarbonylation reactions)
[4][6]. It is therefore not surprising that several important methods have been developed in which a single Pd(II)-based catalytic system promotes, in one synthetic step, the sequential heterocyclization−cyclocarbonylation of suitably functionalized olefinic substrates that carry two nucleophilic moieties placed in appropriate positions to undergo double cyclization.
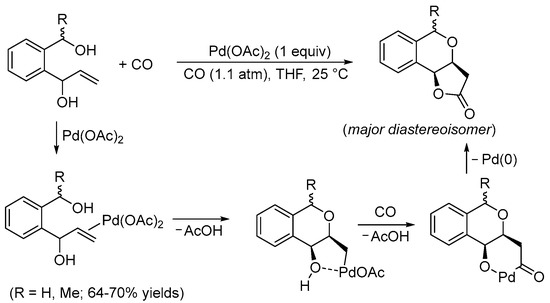
Pioneering studies on this kind of reactivity were conducted by the Semmelhack and Yoshida research groups during the 1980s. In 1984, Semmelhack et al. reported the Pd(II)-promoted stereoselective carbonylative double cyclization of 1-(2-(hydroxymethyl)phenyl)prop-2-en-1-ols to give 3,3a,5,9b-tetrahydro-2
H-furo[3,2-
c]isochromen-2-ones with a
cis junction between the newly formed rings using a stoichiometric amount of Pd(OAc)
2 [5][7]. The process started with the intramolecular 6-
exo-
trig nucleophilic attack of the benzylic hydroxyl group to the double bond, activated by coordination to the Pd(II) center. This led to the formation of a
cis-type alkylpalladium intermediate stabilized by chelation of the second hydroxyl group. The final bicyclic product was then formed through CO migratory insertion followed by intramolecular nucleophilic displacement by the hydroxyl, possibly via the formation of a palladacycle followed by reductive elimination (
Scheme 1).
Scheme 1. Synthesis of 3,3a,5,9b-tetrahydro-2
H-furo[3,2-
c]isochromen-2-ones from 1-(2-(hydroxymethyl)phenyl)prop-2-en-1-ols
[5][7].
Interestingly, when an oxidant for Pd(0) such as CuCl
2 was employed to make the process catalytic, the reaction led to the formation of (
E)-(2-(3-chloroprop-1-en-1-yl)phenyl)methanol from allylic chlorination (74% yield)
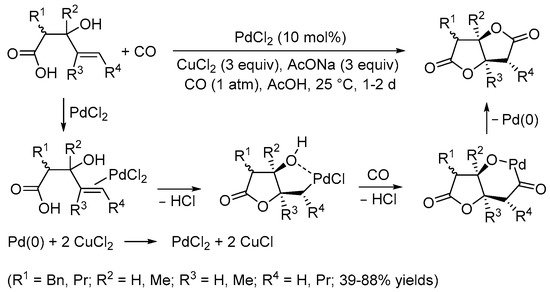
[5][7]. Later on, however, suitable conditions were elaborated by the Yoshida group for performing the carbonylative double cyclization of 3-hydroxy-4-pentenoic acids to stereoselectively give tetrahydrofuro[3,2-
b]furan-2,5-diones with a
cis junction between the rings under Pd(II) catalysis (10 mol% PdCl
2 in the presence of 3 equiv of CuCl
2 and 3 equiv of AcONa, in glacial acetic acid as the solvent, at room temperature and under 1 atm of CO) (
Scheme 2)
[6][8]. The process took place through 5-
exo-
trig cyclization by the intramolecular nucleophilic attack of the carboxylic group to the double bond coordinated to the metal center, stabilized by hydroxyl chelation, to give a
cis-type alkylpalladium complex followed by CO insertion and intramolecular nucleophilic displacement, possibly via the formation of a palladacycle followed by reductive elimination (
Scheme 2)
[6][8].
Scheme 2. Synthesis of tetrahydrofuro[3,2-
b]furan-2,5-diones from 3-hydroxy-4-pentenoic acids
[6][8].
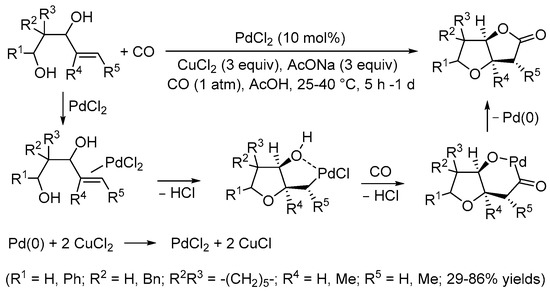
The same research group then published the carbonylative double cyclization of 4-ene-1,3-diols under similar reaction conditions to obtain tetrahydrofuro[3,2-
b]furan-2(3
H)-ones (
Scheme 3)
[7][9].
Scheme 3. Synthesis of tetrahydrofuro[3,2-
b]furan-2(3
H)-ones from 4-ene-1,3-diols
[7][9].
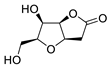
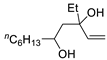
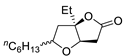
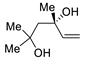
Considering that the bicyclic tetrahydrofuro[3,2-
b]furan-2(3
H)-one substructure is largely found in natural and biologically active molecules, the methods disclosed by Semmelhack and Yoshida for constructing this important core by the carbonylative double cyclization of enediol derivatives have been largely employed as the key step in the semi- or total synthesis of natural products and bioactive compounds. Representative examples are shown in
Table 1.
Table 1. Representative examples of the Pd(II)-promoted carbonylative double cyclization of enediol derivatives in the synthesis of natural and bioactive products.
| Entry |
Conditions |
Substrate |
Product |
Yield (%) |
Refs. |
| 1 |
PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 41 h |
 |
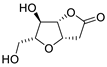 |
63 |
[8][10] |
| 2 |
PdCl2(MeCN)2, (10 mol%), CuCl2 (2.4 equiv), CO (1 atm), THF, 25 °C, 24 h |
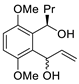 |
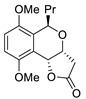 |
65 |
[9][11] |
| 3 |
PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 8 h |
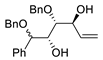 |
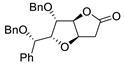 |
85 |
[10][12] |
| 4 |
PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (4 equiv), CO (1 atm), AcOH, 25 °C, 24 h |
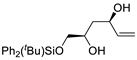 |
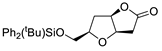 |
93 |
[11][13] |
| 5 |
PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 33 h |
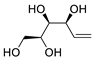 |
 |
38 |
[12][14] |
| 6 |
PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 15 h |
 |
 |
>80 |
[13][15] |
| 7 |
Pd(OAc)2 (1.5 equiv), CO
(1.1 atm), THF, 23 °C, 4 h |
 |
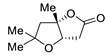 |
87 |
[14][16] |
| 8 |
PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 15 h |
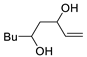 |
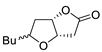 |
81 |
[15][17] |
| 9 |
PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C |
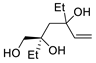 |
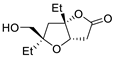 |
63 |
[16][18] |
| 10 |
PdCl2, CuCl, AcONa, CO, AcOH |
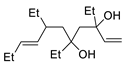 |
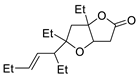 |
33 |
[17][19] |
| 11 |
PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 10 h |
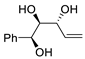 |
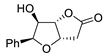 |
85 |
[18][20] |
| 12 |
PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 23 °C, 24 h |
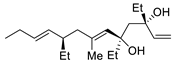 |
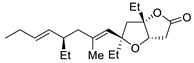 |
75 |
[19][21] |
| 13 |
Pd(OAc)2 (1.5 equiv),
N-methylmorpholine (3 equiv), CO, THF, 25 °C, 15 h |
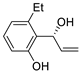 |
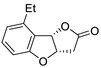 |
58 |
[20][22] |
| 14 |
PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 20 h |
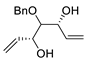 |
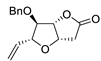 |
65 |
[21][23] |
| 15 |
Pd(OAc)2 (10 mol%), CuCl2
(3 equiv), AcONa (3 equiv),
CO (1 atm), AcOH, 25 °C, 15 h |
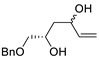 |
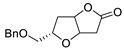 |
63, 70 |
[22][23][24,25] |
| 16 |
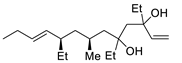
PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 24 h |
 |
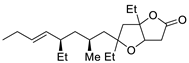 |
33 |
[24][26] |
| 17 |
PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 24 h |
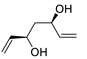 |
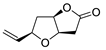 |
87 |
[25][27] |
| 18 |
PdCl2 (10 mol%), CuCl2 (3 equiv), AcONa (3 equiv), CO (1 atm), AcOH, 25 °C, 12 h |
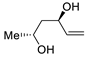 |
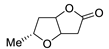 |
61 |
[26][28] |
| 19 |
PdCl2(MeCN)2 (10 mol%), CuCl2 (5 equiv), AcOLi (5 equiv), [Fe(CO)5] (0.5 equiv), AcOH, 60 °C, 1 h |
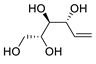 |
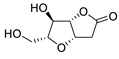 |
47 |
[27][29] |
| 20 |
PdCl2(MeCN)2 (10 mol%), Cu(OAc)2 (4 equiv), LiCl
(4 equiv), [Fe(CO)5] (0.25 equiv), AcOH, 60 °C, 15 min |
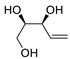 |
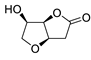 |
67 |
[28][30] |
| 21 |
PdCl2(MeCN)2 (10 mol%), CuCl2 (4 equiv), AcOLi (4 equiv), [Fe(CO)5] (0.3 equiv), AcOH, 60 °C, 30 min |
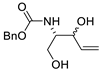 |
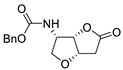 |
75 |
[29][31] |
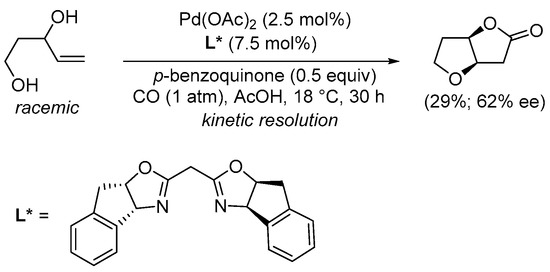
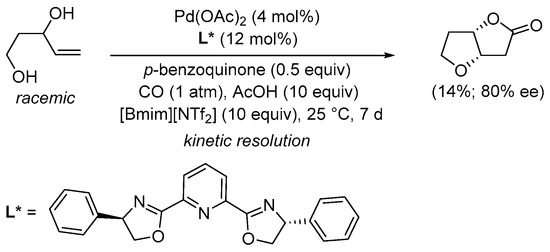
Interestingly, using the appropriate enantiopure ligand, a kinetic resolution of (±)-pent-4-ene-1,3-diols was possible with the formation of the corresponding bicyclic lactone in noracemic form. This was exemplified by the Pd(OAc)
2-catalyzed carbonylation of (±)-pent-4-ene-1,3-diol performed in the presence of an enantiopure bis(oxazoline) ligand and
p-benzoquinone as an external oxidant to give (3a
R,6a
R)-tetrahydrofuro[3,2-
b]furan-2(3
H)-one in 29% yield and 62% ee (
Scheme 4)
[30][32].
Scheme 4. Kinetic resolution of (±)-pent-4-ene-1,3-diol leading to enantioenriched (3a
R,6a
R)-tetrahydrofuro[3,2-
b]furan-2(3
H)-one
[30][32].
More recently, the kinetic resolution of (±)-pent-4-ene-1,3-diols to give nonracemic tetrahydrofuro[3,2-
b]furan-2(3
H)-ones [2-(
S,
S) up to 80% ee, 2-(
R,
R) up to 57% ee] has been realized under similar conditions [4 mol% of Pd(OAc)
2, 12 mol% of 2,6-bis[(4
R)-4-phenyl-2-oxazolinyl]pyridine as enantiopure ligand, 0.5 equiv of
p-benzoquinone, and 10 equiv AcOH] using an ionic liquid as the solvent (such as 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide, [Bmim][NTf
2], 10 equiv), as shown in
SchemeScheme 5 5 [31][33].
Scheme 5. Kinetic resolution of (±)-pent-4-ene-1,3-diol in [bmim][NTf
2] leading to enantioenriched (3a
S,6a
S)-tetrahydrofuro[3,2-
b]furan-2(3
H)-one
[31][33].
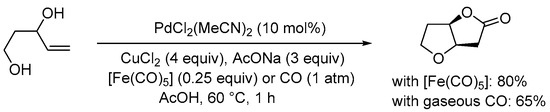
Interestingly, the Gracza group reported that the use of iron pentacarbonyl as an in situ liquid CO source may lead to improved results (significantly shorter reaction times, in particular) in the Pd(II)-catalyzed carbonylative double cyclization of enediols with a terminal double bond, as shown in
SchemeScheme 6 6 [27][32][33][29,34,35].
Scheme 6. Carbonylative double cyclization of pent-4-ene-1,3-diol using [Fe(CO)
5] as in situ CO source
[32][34].
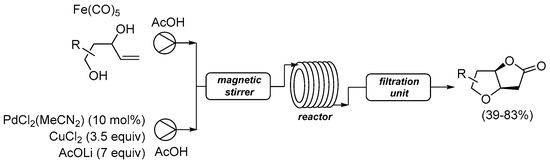
The same research group recently reported their reaction under flow conditions using a continuous microflow system, as shown in
SchemeScheme 7 7 [29][34][31,36].
Scheme 7. Carbonylative double cyclization of 4-ene-1,3-diols using [Fe(CO)
5] as in situ CO source under flow conditions
[34][36].
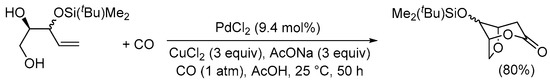
The carbonylative double cyclization process of enediols has also been reported to occur with 4-ene-1,2-diol derivatives. In this case, after the initial 5-
exo-
trig O-cyclization, in the cyclocarbonylation it is the free hydroxyl at C-2 that acts as internal nucleophile, with the formation of a 6-membered ring. This is illustrated by the formation of 8-((
tert-butyldimethylsilyl)oxy)-2,6-dioxabicyclo[3.2.1]octan-3-one from 3-((
tert-butyldimethylsilyl)oxy)pent-4-ene-1,2-diol, as shown in
SchemeScheme 8 8 [35][37].
Scheme 8. 5-
exo-
trig O-cyclization followed by cyclocarbonylation with 6-membered ring closure
[35][37].
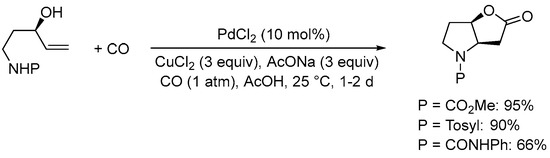
The nucleophilic group undergoing initial heterocyclization can also be nitrogen-based. Thus, as early as 1985 the Tamaru and Yoshida group found that the Pd(II)-catalyzed carbonylative double cyclization of the
N-protected 5-aminopent-1-en-3-ols yielded
N-protected 6-hydroxyhexahydro-2
H-furo[3,2-
b]pyrrol-2-ones, using the same conditions employed for 4-penten-1,3-diols
[36][38]. Among the protective groups tested, the –CO
2Me group turned out as the most suitable, as exemplified in
SchemeScheme 9 9 [37][39].
Scheme 9. Formation of 6-hydroxyhexahydro-2
H-furo[3,2-
b]pyrrol-2-one derivatives from
N-protected 5-aminopent-1-en-3-ols
[37][39].
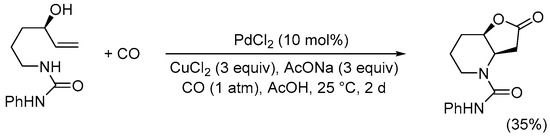
As predicted, owing to the higher degrees of freedom of the alkyl chain,
N-protected 6-aminohex-1-en-3-ols were significantly less reactive, and relatively good results were usually observed with P = CONHPh, as shown in
SchemeScheme 10 10 [37][39].
Scheme 10. Synthesis of 2-oxo-
N-phenylhexahydrofuro[3,2-
b]pyridine-4(2
H)-carboxamide from 1-(4-hydroxyhex-5-en-1-yl)-3-phenylurea
[37][39].
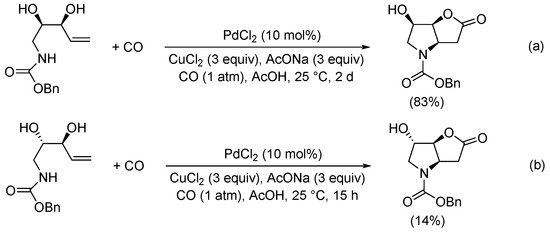
Later on, Jäger et al. reported the carbonylation of benzyl ((2
R,3
S)-2,3-dihydroxypent-4-en-1-yl)carbamate (
Scheme 11a) as a key step in the synthesis of novel 1,4-iminoglycitol derivatives as potential glycosidase inhibitors
[38][40]. On the other hand, the PdCl
2-catalyzed carbonylation of benzyl ((2
S,3
S)-2,3-dihydroxypent-4-en-1-yl)carbamate gave benzyl (3a
R,6
S,6a
S)-6-hydroxy-2-oxohexahydro-4
H-furo[3,2-
b]pyrrole-4-carboxylate in a 14% yield (
Scheme 11b)
[39][41].
Scheme 11. Synthesis of (
a) benzyl (3a
R,6
R,6a
S)-6-hydroxy-2-oxohexahydro-4
H-furo[3,2-
b]pyrrole-4-carboxylate (a precursor for the formation of glycosidase inhibitor derivatives)
[38][40] and (
b) benzyl (3a
R,6
S,6a
S)-6-hydroxy-2-oxohexahydro-4
H-furo[3,2-
b]pyrrole-4-carboxylate
[39][41].
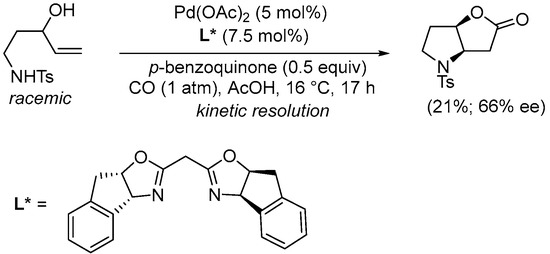
Kinetic resolution of
N-protected 5-aminopent-1-en-3-ols in the Pd(II)-catalyzed carbonylative double cyclization has been reported by Gracza et al. Thus, using enantiopure bisoxazoline ligands, nonracemic hexahydro-2
H-furo[3,2-
b]pyrrol-2-ones could be obtained as in
SchemeScheme 12 12 [40][42].
Scheme 12. Kinetic resolution of (±)-
N-(3-hydroxypent-4-en-1-yl)-4-methylbenzenesulfonamide leading to enantioenriched (3a
R,6a
R)-4-tosylhexahydro-2
H-furo[3,2-
b]pyrrol-2-one
[40][42].
The gaseous CO-free conditions elaborated by the Gracza group for the carbonylation of 4-ene-1,3-diols, involving the use of liquid [Fe(CO)
5] as an in situ CO source (
Scheme 6), have also been successfully employed by the same research team in the Pd(II)-catalyzed carboylative double cyclization of
N-protected 5-aminopent-1-en-3-ols, as in
SchemeScheme 13 13 [32][34].
Scheme 13. Carbonylative double cyclization of
tert-butyl (3-hydroxypent-4-en-1-yl)carbamate using [Fe(CO)
5] as in situ CO source
[32][34].
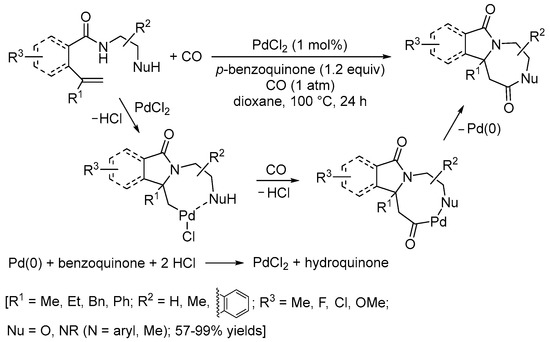
An interesting carbonylative double cyclization process has recently been developed by the Li group
[41][43]. It involved the reaction of
N-(2-aminoethyl)pent-4-enamide or
N-(2-hydroxyethyl)pent-4-enamide derivatives with CO (1 atm) in the presence of PdCl
2 as a catalyst (1 mol%) and
p-benzoquinone as an oxidant (1.2 equiv) (
Scheme 14). As shown in
Scheme 14, the initial 5-
exo-
trig N-cyclization was followed by CO insertion and intramolecular nucleophilic displacement via the formation of an 8-membered ring palladacycle followed by reduction elimination
[41][43].
Scheme 14. Carbonylative double cyclization of
N-(2-aminoethyl)pent-4-enamide and
N-(2-hydroxyethyl)pent-4-enamide derivatives
[41][43].
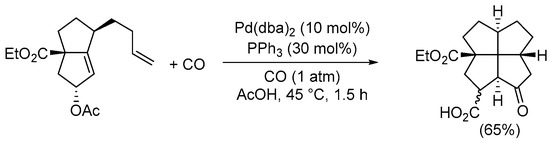
A general approach leading to carbonylative double cyclization is the intramolecular Pauson–Khand reaction starting from suitable diene or enyne substrates. Since several excellent reviews have been published on this reaction
[42][43][44][44,45,46], even in the most recent literature
[45][46][47][47,48,49], this process will not be treated here; however, a particularly striking example in
SchemeScheme 15 15 gives the reader an idea of the powerfulness of this synthetic method for constructing complex carbonylated polycyclic compounds
[48][50].
Scheme 15. Synthesis of 2a-(ethoxycarbonyl)-8-oxododecahydropentaleno[1,6-
cd]pentalene-1-carboxylic acid from ethyl 5-acetoxy-1-(but-3-en-1-yl)-2,3,4,5-tetrahydropentalene-3a(1
H)-carboxylate by Pauson–Khand-type intramolecular reaction
[48][50].