Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Leonid N Maslov and Version 2 by Jason Zhu.
Acute myocardial infarction (AMI) remains the leading cause of mortality in the world, highlighting an urgent need for the development of novel, more effective approaches for the treatment of AMI. Remote postconditioning (RPost) of the heart could be a useful approach. It was demonstrated that RPost triggers infarct size reduction, improves contractile function of the heart in reperfusion, mitigates apoptosis, and stimulates autophagy in animals with coronary artery occlusion and reperfusion. Endogenous opioid peptides and adenosine could be involved in RPost. It was found that kinases and NO-synthase participate in RPost. KATP channels, MPT pore, and STAT3 could be hypothetical end-effectors of RPost.
- heart
- remote postconditioning
- ischemia
1. Introduction
In-hospital mortality in patients with ST-segment elevation myocardial infarction (STEMI) is 5–7% and has not decreased in recent years [1][2][3][1,2,3]. The only effective treatment for acute myocardial infarction (AMI) is recanalization of infarct-related coronary arteries by thrombolysis or percutaneous coronary intervention (PCI). In recent years, the effectiveness of reperfusion therapy has reached its maximum, so in-hospital mortality is not decreasing [1][2][3][1,2,3].
Reperfusion injury to the heart is becoming increasingly relevant in cardiac injury as a result of the increased efficiency of recanalization of infarct-related arteries [4]. There is a clear urgent need to develop new approaches to prevent cardiac reperfusion injury. Remote postconditioning (RPost) could be such an approach. Remote postconditioning is the increased tolerance of an organ to reperfusion after long-term ischemia as a result of the short-term ischemia/reperfusion (I/R) of a remote organ.
2. Experimental Data
2.1. The Involvement of the Nervous System in the Mechanism of Remote Postconditioning
Rats underwent coronary artery occlusion (CAO, 30 min) and reperfusion (2 h) [5][16]. RPost was mimicked by vagal stimulation. Stimulation of n. vagus decreased the infarct size [5][16]. In another study, rats were subjected to CAO (30 min) and reperfusion (2 h) [6]. RPost was triggered by a 15 min occlusion of femoral arteries. RP induced infarct size reduction by 56%. Vagotomy or the denervation of hind limbs completely abolished the cardioprotection triggered by RPost. Investigators concluded that the peripheral nervous system is involved in the cardioprotective effect of RPost [6]. However, sensory nerves probably did not participate in RPost because the activation of the transient receptor potential cation channel-1 (TRPV1 channel) localized on sensor nerves by capsaicin 5 min before reperfusion did not affect infarct size [6]. Mice underwent CAO (45 min) and reperfusion (24 h) [7][17]. RPost was mimicked by a 2 cm transverse incision to the abdominal midline at the end of CAO. Nociceptive RPost caused a decrease in infarct size by 72%. Investigators concluded that skin nociceptors could be involved in trauma-induced cardioprotection [7][17]. These findings are questionable because the thoracotomy that precedes the CAO is itself a strong trauma. In addition, animals were anesthetized by pentobarbital which contributed to a decrease in pain sensitivity. Mice underwent CAO (45 min) and reperfusion (4 or 24 h) [8][18]. Nociception was induced during reperfusion by electrical stimulation of needles inserted into the skeletal muscle. Lidocaine was injected subcutaneously to block skin sensory nerves, Nociceptive postconditioning reduced infarct size by about 60%. Lidocaine eliminated nociceptive postconditioning. The infarct-sparing effect of nociceptive-induced postconditioning was not found in TRPV1-channel knockout mice [8][18]. These findings indirectly indicate that the nervous system and sensory nerves could be involved in RPost. However, the significance of the results was questionable because investigators used small groups of mice (n = 6) [8][18]. Rats were subjected to CAO (30 min) and reperfusion (3 h) [9][19]. RPost was performed by three cycles of the occlusion (5 min) and reperfusion (5 min) of both femoral arteries using clamps immediately after the onset of reperfusion. It was found the TRPV1 channel blocker capsazepine completely reversed the infarct-limiting effect of RPost in rats with CAO (30 min) and reperfusion [9][19]. CGRP8-37, a calcitonin gene-related peptide (CGRP) receptor antagonist (2 mg/kg intravenously, 2 min before reperfusion), also abolished RPost-induced cardiac tolerance to reperfusion. RPost increased the CGRP level in the heart and plasma [9][19]. Consequently, the TRPV1 channel and CGRP are involved in RPost-triggered cardioprotection. These findings demonstrated that the nervous system, the autonomic nervous system, the TRPV1 channel, CGRP, and α7nAChR receptor could be involved in the cardioprotective effect of RPost (Figure 1).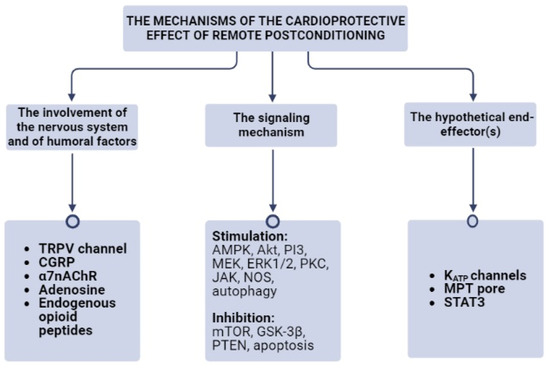
Figure 1. The mechanisms of the cardioprotective effect of remote postconditioning. α7nAChR, the alpha-7 nicotinic receptor; Akt, protein kinase B; AMPK, AMP activated protein kinase; CGRP, the calcitonin gene-related peptide; ERK1/2, extracellular regulated kinase; GSK-3β, glycogen synthase kinase-3β; JAK, janus kinase; KATP channels, ATP-sensitive K+ channels; MEK, mitogen-activated protein kinase kinase; MPT pore, mitochondrial permeability transition pore; mTOR, mammalian target of rapamycin; NOS, NO-synthase; PI3, phosphoinositide-3-kinase; PKC, protein kinase C; PTEN, phosphatase and tensin homolog; STAT3, signal transducer and activator of transcription 3; TRPV, receptor potential cation channel.
2.2. The Involvement of Humoral Factors in the Mechanism of Remote Postconditioning
Rats were subjected to CAO (30 min) and reperfusion (180 min) [10][5]. RPost was induced by renal artery occlusion for 5 min and reperfusion before the restoration of coronary blood flow. The non-selective adenosine receptor antagonist 8-sulfophenyl theophylline (8-SPT, 10 mg/kg) was injected 5 min before reperfusion. It was found that RPost triggered a decrease in infarct size by about 50%. This effect was completely reversed by 8-SPT. Permanent renal artery occlusion had no effect on infarct size [10][5]. These data demonstrate that some cardioprotective substance is released from the kidney and protects the heart against reperfusion. Investigators did not detect adenosine levels in blood [10][5]. It was reported that the half-life of adenosine in blood is 10 s [11][22]. Therefore, signal transmission from kidneys to the heart via adenosine is questionable. It is probably more likely that adenosine acts on the heart’s level. However, the possibility that RPost induced the long-term release of adenosine from the kidney cannot be ruled out. The isolated rat heart was subjected to three cycles of global ischemia (3 min) and reperfusion (5 min) [12][23]. The coronary effluent was collected during reperfusion. The isolated rat heart was subjected to CAO (30 min) and reperfusion (120 min), and the heart was perfused by the coronary effluent after the onset of reperfusion three times for 30 s (total 90 s). This impact caused a decrease in infarct size by about 50% [12][23]. Investigators could isolate the hydrophobic fraction of the coronary effluent which can mimic RPost. This fraction contained proteins with a molecular weight < 10 kDa [12][23]. Hydrophobic peptide molecules can penetrate the blood–brain barrier [13][24]. It was reported that RPost can protect the brain against I/R [14][15][16][25,26,27]. These data indirectly confirm the hydrophobic nature of molecules involved in RPost. RPost was induced by ischemia (four cycles for 5 min) and reperfusion (three cycles for 5 min) of the hindlimb in pigs [17][28]. The plasma of postconditioned pigs was used for perfusion of the isolated rat heart subjected to global ischemia (30 min) and reperfusion (120 min). Pig plasma reduced the infarct size by 34% [17][28]. This evidence demonstrates the involvement of non-identified humoral factor(s) in RPost-induced cardioprotection. Mice underwent CAO (45 min) and reperfusion (2 h) [7][17]. RPost was mimicked by an abdominal incision in mice anesthetized by pentobarbital. Administration of the bradykinin-2 receptor antagonist Hoe 140 completely abolished cardioprotection induced by a painful stimulus. Bradykinin-2 receptor knockout also eliminated the infarct-reduced effect of nociceptive postconditioning. The β-adrenergic receptor blocker propranolol (2 mg/kg intravenously) also reversed nociceptive postconditioning [7][17]. In 2016, Koyama et al. found that the intracoronary administration of Ringer’s solution containing lactate did not aggravate I/R cardiac injury in patients with STEMI and PCI [18][32]. Investigators proposed that lactate can mimic RPost [19][20][33,34]. Recently, it has been demonstrated that exogenous lactate can mimic RPost in rats [21][35]. It is possible that lactate released from skeletal muscle can trigger RPost in limb I/R. These data indicate that endogenous opioid peptides, CGRP, lactate, and adenosine could be involved in RPost (Figure 1). Non-identified hydrophobic peptide molecules can also participate in RPost. The involvement of bradykinin or catecholamines in RPost needs to be re-examined.3. The Signaling Mechanism of Remote Postconditioning
It was reported that kinases are involved in ischemic preconditioning and postconditioning [22][23][36,37], and are also involved in RPost [24][38].3.1. AMPK and mTOR
It was reported that AMP-activated protein kinase (AMPK) participates in RPost [25][11]. Rats underwent permanent CAO. RPost was performed by three cycles of left forelimb ischemia (5 min) and reperfusion (5 min), and was repeated every day for 3 days. RPost caused a decrease in infarct size by about 30% and inhibited apoptotic cell death in the heart. RPost reduced the phosphorylated mammalian target of rapamycin (p-mTOR) level [25][11]. This kinase is an endogenous inhibitor of autophagy [26][39]. It was found that RPost resulted in an increase in the marker levels of autophagy in myocardial tissue (Beclin-1 and LC3 II), and caused an increase in their p-AMPK content. Pretreatment with the AMPK inhibitor compound C abolished the infarct-limiting effect of RPost [25][11]. Consequently, RPost stimulates AMPK and autophagy, and inhibits mTOR and apoptosis (Figure 1).3.2. Akt, ERK, and PI3 Kinase
RPost was performed by three cycles of right hindlimb I/R in rats with CAO (45 min) and reperfusion (180 min) [27][40]. RPost decreased infarct size by 35% and increased p-Akt levels in the myocardium. Wortmannin, a phosphatidylinositol 3-kinase (PI3-kinase) inhibitor, reversed the increase in p-Akt content in myocardial tissue [27][40]. RPost was induced by four cycles of ischemia (5 min) and reperfusion (5 min) in pigs [17][28], and reduced infarct size by 33%. The plasma of postconditioned pigs was used for perfusion of the isolated rat heart subjected to global ischemia (30 min) and reperfusion (120 min). Pig plasma reduced infarct size by 34% [17][28]. This cardioprotective effect of pig plasma was abolished by wortmannin, a phosphatidylinositol 3-kinase (PI3-kinase) inhibitor, and U0126, a mitogen-activated protein kinase (MEK) inhibitor. U0126 simultaneously inhibits the extracellular signal-regulated kinase-1/2 (ERK1/2)-localized downstream of MEK [17][28]. It was reported that RPost induced by three cycles of hindlimb ischemia (5 min) and reperfusion (5 min) resulted in an increase in the p-Akt level in murine myocardium [28][41]. Mice were subjected to CAO (45 min) and reperfusion (120 min) [29][14]. RPost was carried out by three cycles of unilateral hindlimb ischemia (5 min) and reperfusion (5 min). It decreased infarct size by about 30%, and increased the p-Akt kinase level in myocardial tissue. The PI3-kinase inhibitor LY294002 abolished an increase in the p-Akt kinase level. Rats were subjected to CAO (45 min) and reperfusion (120 min) [30][7]. RPost was performed by three cycles of ischemia (5 min) and reperfusion (5 min) of both hindlimbs, and reduced infarct size by about 50%. The PI3-kinase inhibitor wortmannin abolished the infarct-limiting effect of RPost [30][7]. Thus, PI3-kinase, Akt, MEK, ERK1/2 are involved in RPost (Figure 1).3.3. Protein Kinase C (PKC)
It was found that the PKC inhibitor chelerythrine completely reversed the infarct-reducing effect of RPost triggered by three cycles of bilateral hindlimb I/R in rats with CAO (45 min) and reperfusion (120 min) [30][7]. Consequently, PKC is involved in RPost (Figure 1).3.4. JAK
Mice underwent CAO (45 min) and reperfusion (120 min) [28][41]. RPost was carried out by three cycles of ischemia (5 min) and reperfusion (5 min) of the left hind limb, and it limited infarct size by 44% and inhibited apoptosis. The Janus kinase (JAK) inhibitor AG490 reversed the infarct-reducing effect RPost [28][41]. Thus, JAK participates in RPost (Figure 1).3.5. JNK and GSK-3β
The inhibition of glycogen synthase kinase-3β (GSK-3β) or c-Jun N-terminal kinase (JNK) promoted an increase in cardiac tolerance to I/R [31][32][42,43]. Rats underwent CAO (30 min) and reperfusion (120 min) [33][21]. RPost was performed by bilateral hind limb ischemia (10 min) and following reperfusion, and limited infarct size by about 19%. RPost stimulated the phosphorylation (inactivation) of GSK-3β [33][21], and increased the p-GSK-3β levels in myocardial tissue in mice [34][44]. Limb RPost reduced infarct size, the serum cTroponin I, and creatine kinase-MB levels in rats [33][21]. This cardioprotective effect was associated with an increase in phosphorylated GSK-3β content in myocardial tissue. Thus, GSK-3β inhibition is involved in RPost (Figure 1). The role of JNK inhibition in the cardioprotective effect of RPost has not been studied before.3.6. PTEN
Phosphatase and Tensin Homolog (PTEN) catalyzes kinase dephosphorylation [35][45]. Mice were subjected to CAO (40 min) and reperfusion (24 h) [32][43]. RPost was induced by four cycles of occlusion (5 min) and reperfusion (5 min) of the femoral artery. RPost limited infarct size by 40.7% and reduced the number of apoptotic cells in myocardial tissue. RPost decreased the PTEN level and increased the p-Akt and p-GSK-3β levels in myocardial tissue [34][44]. Mice were fed a 2% cholesterol diet for 12 weeks. This diet abolished RPost-induced cardioprotection, reduced p-Akt and p-GSK-3β levels, and increased the PTEN content in myocardial tissue compared to the RPost + CAO group. The PTEN inhibitor bisperoxovanadium (1.0 mg/kg) restored the cardioprotective effect of RPost in mice with a cholesterol diet [34][44]. It is possible that the activation of PTEN in mice with a cholesterol diet is a reason for the disappearance of RPost-induced cardiac tolerance to I/R. Consequently, PTEN inhibition could be involved in RPost (Figure 1).3.7. NO-Synthase
NO-synthase (NOS) is involved in the cardioprotective effect of the second window of ischemic preconditioning [36][46]. Rabbits were subjected to CAO (30 min) and reperfusion (3 h) [37][9]. RPost was carried out by occlusion (5 min) of the left pulmonary artery following reperfusion (5 min). Investigators did not evaluate infarct size. RPost resulted in a decrease in the plasma concentration of creatine kinase and malondialdehyde (MDA). These findings indicate that RPost can mitigate excessive reactive oxygen species (ROS) production. Pretreatment with the NOS inhibitor L-NAME abolished the cardioprotective effect of RPost [37][9]. It was found that limb RPost triggered an increase in endothelial NOS expression in myocardial tissue of rabbits [38][47]. It was reported that RPost induced by three cycles of hindlimb ischemia (5 min) and reperfusion (5 min) resulted in an increase in the p-eNOS level in the murine myocardium [28][41].4. The Hypothetical End-Effector(s) of Remote Postconditioning
The transcription factor signal transducer and activator of transcription 3 (STAT3) plays an important role in ischemic preconditioning and postconditioning [23][37]. It was found that RPost induced by three cycles of hindlimb ischemia (5 min) and reperfusion (5 min) resulted in an increase in the p-STAT level in murine myocardium [28][41]. Pigs underwent CAO (55 min) and reperfusion (120 min) [39][48]. RPost was induced by tightening a tourniquet around the left hindlimb, and four cycles of ischemia (5 min) and reperfusion (5 min) was used. RPost limited infarct size by 51% and increased the p-STAT level [39][48]. It should be noted that both groups of investigators did not use the STAT inhibitor static. Consequently, there is only indirect evidence of the involvement of STAT3 in RPost. It was reported that ATP-sensitive K+ channels (KATP channels), big conductance Ca2+ channels (BKCa channels), and mitochondrial permeability transition pore (MPT pore) participate in the infarct-limiting effect of pre- and postconditioning [22][23][40][36,37,49]. Thus, it is possible that KATP channels, the MPT pore, and STAT3 could be end-effectors of RPost (Figure 1).5. Remote Postconditioning and Experimental Metabolic Syndrome
Researchers already reported above that a long-term 2% cholesterol diet reversed the cardioprotective effect of RPost [34][44]. Researchers induced metabolic syndrome (MS) in rats with a high-carbohydrate high-fat diet (90 days) [41][50]. Rats were subjected to CAO (45 min) and reperfusion (120 min). RPost was induced by three cycles of bilateral hindlimb ischemia (5 min) and reperfusion (5 min). The infarct-limiting effect of RPost in young (150 days) rats with MS was less than in animals without MS (p < 0.001) [39][48]. RPost did not reduce infarct size in aged rats (540 days) with MS [42][20]. Researchers found a direct correlation between infarct size and the plasma leptin level (r = 0.85, p < 0.014) in rats with RPost and MS [42][43][20,51]. It was reported that leptin increased cardiac tolerance to I/R in rats through the activation of the JAK2/STAT3 signaling pathway where JAK is Janus kinase, and STAT is the signal transducer and activator of transcription [44][52]. It could be proposed that an increase in the endogenous leptin level will enhance cardiac tolerance to I/R but Researchers found that an increase in the plasma leptin concentration is associated with a rise in infarct size [42][20].6. The Optimal Protocol of Remote Postconditioning
Three cycles of bilateral hindlimb I/R promoted infarct size reduction by 36–56% [6][9][30][6,7,19]. Single bilateral hindlimb ischemia contributed to a decrease in infarct size by about 19% [33][21]. Three or four cycles of unilateral limb I/R promoted infarct size reduction by 30–51% [17][25][28][34][39][11,28,41,44,48]. Most investigators used three or four cycles of unilateral hindlimb I/R [17][28][34][39][28,41,44,48]. Xu et al. (2022) carried out RPost by three cycles of left forelimb ischemia (5 min) and reperfusion (5 min). In this case, RPost caused a decrease of in infarct size by about 30%, Researchers suggest that the efficiency of RPost depends on the skeletal muscle volume involved in RPost. The larger volume promoted the small infarct size. Single I/R of extremities exhibits a weak infarct-reducing effect. Consequently, three or four cycles of I/R on hindlimbs should be used for an increase in cardiac tolerance to I/R (Figure 2).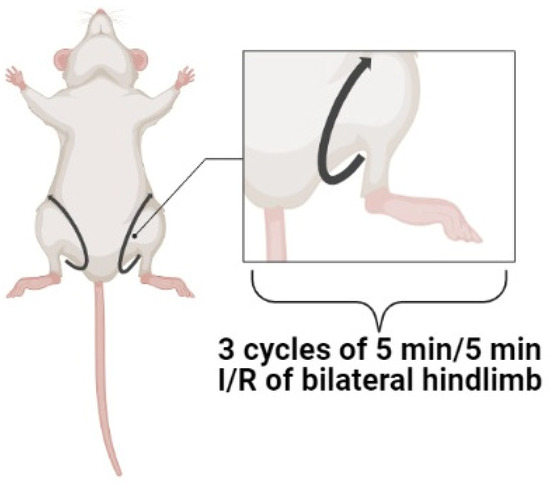
Figure 2. The optimal protocol of remote postconditioning. I/R, ischemia/reperfusion.