2. Retina
The retina transduces and preprocesses the information via an intraretinal circuit and sends it via the optic nerve to the visual nuclei of the brain
[29][43]. The mature vertebrate retina is organized into layers
[30][31][44,45], such as (i) the outer nuclear layer (ONL), composed of the cell bodies of rod and cone photoreceptors, located closest to the retinal pigmented epithelium (RPE); (ii) the inner nuclear layer (INL), composed of the cell bodies of interneurons-horizontal, bipolar, amacrine, and displaced ganglion cells-located between the ONL; and (iii) the ganglion cell layer (GCL). The GCL is located near the vitreous body of the retina and consists of ganglion cells, displaced amacrine cells, and some light-sensitive ganglion cells. Interspersed between these nuclear layers are the outer plexiform layer (OPL) and the inner plexiform layer (IPL), where synaptic contacts occur. Ganglion cells are the projection neurons, and their axons form the nerve fiber layer (NFL) within the retina and give rise to the optic nerve as they exit the retina
[30][32][44,46].
Photoreceptors communicate with the rod- or cone-specific bipolar cells and horizontal cells in the OPL. Bipolar cells synapse with ganglion cells in the IPL. This circuit is named the vertical/radial pathway, and all cells use glutamate as a neurotransmitter
[30][33][44,47]. Horizontal and amacrine cells may modulate the radial pathway in the OPL and IPL and mediate the processing of light/image stimuli, contrast, and detection of movement direction
[34][48].
The retinal vasculature consists of the choroidal and intraretinal blood vessels responsible for the blood supply of the outer and inner retina, respectively. In humans, the inner retina presents a peripapillary plexus in the innermost part of the NFL, accompanied by inner (in the GCL) and outer (in the IPL, INL to OPL) intraretinal beds. Interestingly, the specialized region for high visual acuity in primates, the fovea, is avascular, which minimizes light distortion
[35][49].
In addition to neurons, the retina contains three types of glial cells, Müller cells, astrocytes, and microglia. Müller glia, which comprises 90% of retinal glial cells, have a cell body in the center of the INL and radial processes that extend through all retina layers. Consistent with the multiple roles that Müller glia play in retinal homeostasis and physiology, dysfunction of these cells in DR may be related to retinal dysfunction and severity of DR
[33][34][36][37][47,48,50,51].
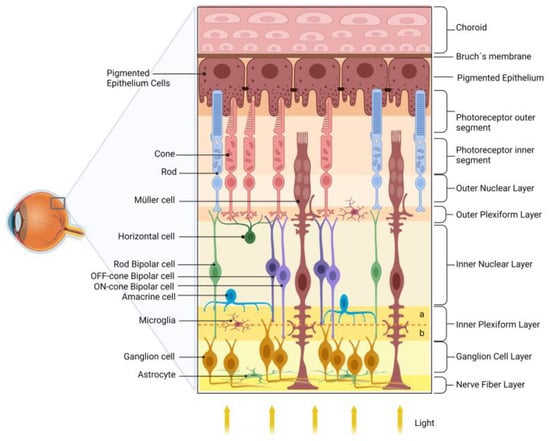
Figure 1 shows a schematic drawing of the retina.
Figure 1. Schematic diagram of the retina. The retina is organized into nuclear layers separated by plexiform layers. The photoreceptors (rods and cones) have nuclei in the outer nuclear layer and synapse with bipolar and horizontal cells in the outer plexiform layer. Bipolar cells are divided into rod bipolar (light green) and cone bipolar (light and dark purple). Cone bipolar cells form synapses directly with their respective ganglion cells in specific sublamina (a and b, for, respectively, OFF and ON pathways) of the inner plexiform layer (IPL). Rod bipolar indirectly transmits information to ganglion cells indirectly via an amacrine cell in the IPL. The ganglion cell layer and the nerve fiber layer are formed by the nuclei and axons of ganglion cells, respectively, which transmit the information to the visual encephalic centers. The retinal glial cells are Müller cells, astrocytes, and microglia. The retinal pigmented epithelium (RPE), composed of pigmented epithelial cells, plays several essential roles in maintaining the retina’s health.
3. Diabetic Retinopathy
DR is a multifactorial pathology that results from a dysfunction in a variety of retinal cells, including Müller glia, ganglion cells, endothelial cells, and photoreceptors, as well as optic nerve damage and, later, retinal neuronal cell death and vascular lesions
[38][39][52,53]. Although neural retina/optic nerve changes occur before vascular alterations in both human and animal models, DR is a chronic microvascular disease characterized by vasculopathy, hyperpermeability, hypoperfusion, and neoangiogenesis of slow progression, accompanied by vitreous hemorrhage, retinal detachment, diabetic macular edema process (DME), and/or macular ischemia, resulting in neural abnormalities and retinal deficits
[20][40][41][20,54,55]. Although DME and macular ischemia can occur at any stage of the disease, they are more common in the later stages of DR
[40][54].
Chronic hyperglycemia promotes the thickening of the retinal capillary basement membrane, impairs adherence of tight junctions between endothelial cells, and makes blood vessels more permeable. These events define the process known as the blood–retinal barrier (BRB) breakdown. This phenomenon is accompanied by decreased pericytes and mesenchymal cells surrounding endothelial cells, providing structural support for the vessel wall and vascular repair processes
[42][43][56,57]. The loss of pericytes leads to voids in the capillary walls. In response, capillary endothelial cell growth is induced toward the inner part of the vessel wall, favoring the formation of cell clusters that obstruct the vessel lumen and the appearance of hemorrhagic spots and microaneurysms. This combination of damage compromises vascular function and the oxygen and nutrient supply to the retinal tissue, leading to ischemia, increased synthesis, and release of pro-angiogenic factors and neovascularization
[41][43][55,57].
DR is classified based on pathophysiological characteristics and vascular manifestations during the clinical course, the initial phase, known as non-proliferative DR (NPDR), and the advanced phase, known as proliferative DR (PDR). The presence of new blood vessels characterizes the PDR stage
[44][58].
NPDR is characterized by morphological changes and vascular damage, including alterations in the microvasculature and an increase in vascular caliber. During this phase, hyperglycemia causes hemodynamic dysfunction, increased permeability, microaneurysms, hemorrhages, and exudates. These manifestations generally progress slowly, and some patients may be asymptomatic. As the disease progresses, the frequency of hemorrhagic events increases, disrupting the retina’s oxygen supply and metabolic demands and increasing the risk of ischemic events. Fundus examination may reveal whitish spots known as “cotton-wool spots” that indicate ischemic processes or retinal infarction
[42][45][46][56,59,60]. Thus, the abnormal angiogenic process leads to a more advanced and critical PDR
[41][47][55,61].
Approximately half of NPDR patients progress to PDR in less than one year
[48][62]. The main feature of PDR is the appearance of new blood vessels because of the local accumulation of vascular growth factors caused by prolonged hypoxia, followed by the formation of ischemic zones. These vessels are delicate, composed entirely of the endothelium, and grow along the retina and into the vitreous without initially causing symptoms or vision loss. However, because their walls are thin and fragile, the vessels can rupture and release blood into the vitreous cavity, causing severe vision loss and even blindness
[41][49][55,63]. Neovascularization is usually accompanied by fibrous tissue growth, which is visible on ophthalmoscopy
[48][62].
Although DR has been predominantly considered a microvascular disease, increasing evidence suggests that retinal dysfunction and degeneration are critical features associated with disease progression and that early neural injury occurs before vascular damage in DR
[50][64]. Data from electroretinograms of diabetic patients show a decrease in the oscillatory potential, which would be the first observable change in the retina of these individuals
[51][65]. Other parameters are altered in individuals with DM without vascular changes, such as contrast sensitivity, color vision, and oscillatory potentials
[39][53]. Histologic analysis of clinical and postmortem specimens confirmed alterations in retinal structural parameters, such as changes in the thickness of different retinal layers, neuronal death, and increased oxidative stress before vascular changes visible with fundoscopy, supporting the concept of an early neurodegenerative phase of DR preceding NPDR
[51][52][53][54][65,66,67,68].
In this sense, a prospective and longitudinal human study of 45 patients with type 1 DM, without or with minimal DR, reported significant and progressive impairment of both the nerve fiber layer (NFL) and the ganglion cell (GC)/inner plexiform layer, independent of glycated hemoglobin, age, and sex
[50][64]. To confirm these findings, eye samples from six deceased patients with DM, without or with minimal DR, were compared to eyes from donors of a similar age without DM. The first group showed a significantly thinner NFL but no difference in GC density. Surprisingly, the data showed that despite the neuronal changes observed in the DR group, there was no difference in retinal capillary density between the groups. These findings were confirmed in two experimental diabetes mouse models. Recently, other studies using optical coherence tomography showed decreased retinal thickness (INL/GCL and NFL) in diabetic patients without signs of DR, indicating neurodegeneration before vascular changes
[50][55][56][57][58][64,69,70,71,72]. These results suggest that retinal diabetic neuropathy is not necessarily a consequence of retinal ischemia. It develops gradually, independent of glycated hemoglobin, age, and sex, before the onset of microvasculopathy.
All retinal cells (e.g., ganglion, bipolar, amacrine, and photoreceptor cells) show altered function before visible microvascular lesions
[39][53]. Therefore, the description of DR as a mere “microvascular pathology” is an oversimplified and flawed characterization that hinders the exploration of novel therapeutic approaches based, for example, on intervention on neurodegenerative pathways
[59][73].
4. Cellular and Molecular Pathways Involved with Oxidative Stress and Inflammatory Response during DR
The retina is susceptible to oxidative stress due to its high polyunsaturated fatty acids (PUFA) concentration and rapid oxygen and glucose uptake. Another critical element in the pathophysiology of DR is inflammation. The oxidative stress induces ROS production, the root cause of neuropathy and retinopathy
[60][74].
Glucose is the primary precursor for energy production in the cell. The citric acid cycle requires glycolysis-derived pyruvate to produce the high-energy electron carriers FADH
2 and NADH, which fuel the electron transport required for ATP production. At the end of this electron transport chain, oxygen molecules serve as electron acceptors, limiting reactions that would otherwise impair the function of other cellular components. High glucose levels cause increased glycolysis rate, pyruvate levels, and FADH
2 and NADH production. When these electron carriers reach the upper limit of the voltage gradient across the inner mitochondrial membrane, electron transfer through complex III is impaired, pushing electrons back to coenzyme Q, which mediates superoxide (O
2−) production
[61][75]. Superoxide belongs to a class of reactive oxygen species (ROS). ROS are highly reactive, mainly due to unpaired electrons in the outer shell of their atoms, causing dysfunction in enzymes and altering the interaction properties of proteins, lipids, sugars, and nucleic acids. O
2− can be converted by superoxide dismutase activity to the non-radical ROS, hydrogen peroxide (H
2O
2). H
2O
2 alters protein interactions or function through cysteine oxidation. H
2O
2 can be converted to hydroxyl radical (OH
.) by the Fenton reaction, as well as to hypochlorous acid (HOCl) when targeted by neutrophil myeloperoxidase during an inflammatory process
[62][63][76,77]. Indeed, diabetes-induced neuroinflammation showed increased NO synthase (NOS2) levels in the retina. The reaction of nitric oxide (NO) with O
2− forms the family member of the reactive nitrogen species peroxynitrite (ONOO−). ROS damages DNA leading to the activation of DNA repair enzymes, such as poly (adenosine diphosphate (ADP)-ribose) polymerase. This enzyme produces ADP-ribose, which inhibits glycerol-3-phosphate dehydrogenase (GAPDH), which is responsible for a step in the glycolytic pathway. The result is an accumulation of glycolytic intermediates used by the polyol and hexosamine pathways, advanced glycation end products (AGEs), and upregulation of the PKC signaling pathway
[64][78].
All these pathways increase ROS and form a positive feedback loop to increase oxidative stress in the cell. Among many ROS-generating systems, NADPH oxidase (NOX) activity seems particularly important for generating cytoplasmic ROS, especially for neovascularization
[65][79]. NOX is a group of enzymes that catalyzes the production of O
2− by transferring an electron from NADPH to O
2. NOX4, a subtype of NOX, can also produce H
2O
2. NOX activity is upregulated by several signals produced during the progression of DR, including tumor necrosis factor-alpha (TNF-α), several interleukins, and AGE signaling. Therefore, NOX appears to be important in advance of DR, and inhibition of NOX has emerged as a promising therapeutic target for DR
[66][67][80,81]. Thus, oxidative stress is a constitutive problem and a hallmark of DM.
As mentioned above, hyperglycemic conditions induce the production of AGEs. Briefly, AGEs are generated by the reduction in amino acids, lipids, or nucleic acids by glucose, triggering the formation of Schiff bases. Rearrangements of Schiff bases lead to forming Amadori products, which have reactive carbonyl groups. These carbonyl groups interact with amino, sulfhydryl, and guanidine functional groups, denaturing and cross-linking target proteins. Finally, reactions of Amadori products with arginine or lysine within target proteins lead to the formation of stable AGEs. For example, AGE can signal through specific receptors, the receptor for advanced glycation end products (RAGE), mediating paracrine and autocrine signaling involved in the pathology of diabetic retinopathy
[68][82].
AGEs and oxidative stress activate retinal micro- and macroglia during diabetic retinopathy
[69][83]. Once activated, microglia produced interleukin (IL)-1β, IL-6, IL-8, and TNFα under the control of NF-kappaB (NF-kB)
[70][71][72][73][74][75][84,85,86,87,88,89]. Müller glia is activated in DR, leading to increased secretion of IL-6, IL-8, VEGF, INF-γ, TNF-α, IL-1β, monocyte chemoattractant protein-1 (MCP-1), and EGF
[76][77][90,91]. In addition, galectin-3 is upregulated in Müller glia in diabetic rats, and the absence of galectin-3 reduces the number of astrocytes and macrophage-like cells, both quiescent and activated, in the optic nerve and protects against ganglion cell death
[78][92]. Retinal astrocytes are consistently activated by oxidative damage, increasing their secretion of IL-6, MCP-1, and MIP-2 under the control of p38MAPK and NF-kB signaling
[79][93]. In addition, the AGE receptor mediates astrocyte activation biased toward a pro-inflammatory profile, increasing NF-kB and complement C3 expression, mediating synaptic loss
[80][94].
All these secreted mediators orchestrate various pathological events leading to persistent inflammation, ROS production, neurodegeneration, blood–retinal barrier permeability, and defective neovascularization, which are hallmarks of diabetic retinopathy. TNF-α and galectin-3 mediate apoptotic cell death of retinal ganglion cells
[78][81][92,95]. Both IL-1β and TNF-α increased the content of ICAM-1 on the endothelial surface, allowing monocyte diapedesis through VLA4 and CD18 binding
[82][96]. These monocytes are attracted to the retina by MCP-1 secreted by astrocytes and Müller glia and differentiate into macrophages once in the retinal tissue
[83][97]. IL-8 recruits neutrophils, which release myeloperoxidase granules, further contributing to the maintenance of oxidative stress
[84][85][98,99]. These leukocyte recruitments rely on the luminal exposure of MIP-2, which stimulates the upregulation of leukocyte integrins during diapedesis
[86][100]. Oxidative damage and IL-6 stimulate VEGF production by Müller glia and endothelial cells, respectively
[87][101]. VEGF decreases occludin content in endothelial cells, thereby increasing the permeability of the blood–retinal barrier, leading to retinal edema and degeneration
[88][102]. To worsen the scenario, VEGF and EGF stimulate neovascularization, expanding the area covered by defective capillaries in the proliferative phase of diabetic retinopathy
[89][103]. Thus, neuroinflammation and oxidative stress produce each other in a positive feedback loop, contributing to neuronal death and vascular pathology of diabetic retinopathy
[39][53].