Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 7 by Jessie Wu and Version 6 by Jessie Wu.
Autophagy is a highly conserved cellular degradation process that regulates cellular metabolism and homeostasis under normal and pathophysiological conditions. Autophagy and metabolism are linked in the hematopoietic system, playing a critical role in the self-renewal, survival and differentiation of hematopoietic stem and progenitor cells, and in cell death, particularly influencing the cell fate of the hematopoietic stem cell pool. In leukemia, autophagy supports leukemia cell growth, contributes to leukemia stem cell survival and resistance to chemotherapy. Acute myeloid leukemia (AML), a common type of acute leukemia with poor survival and prognosis,
- autophagy
- hematopoiesis
- acute myeloid leukemia
- metabolism
- therapy resistance
1. Autophagy and Acute Myeloid LeukemiaL Stem Cells
During leukemic development, LSCs can adapt their metabolism and autophagyic mechanisms to supplyprovide the high energy and nutrients required for LSCs proliferation and survival under conditions of nutrient deficiency, starvation, hypoxia, or during chemotherapy treatments [1][2][3][4][5][7 , 91 , 92 , 93 , 94 ] .
Spartecificular, theally, adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK), a protein complex fundamentcritical in mitochondrial metabolism and mitophagy, is constitutively activated in LSCs, increasing mitochondrial clearance to support LSCs growth and survival through its downstream target FIS1, the mitochondrial fission 1 protein component of a mitochondrial complex that promotes mitochondrial fission [7][ 96 ]. AMPK/FIS1-mediated mitophagy is required for theLSC self-renewal and survival of[ LSCs [7]96]. An oOverexpression of FIS1 was also found in AML cells while FIS1 depletion of FIS1 impairs mitophagy, weakening the self-renewal capacity of LSCs and determiningresulting in the induction of myeloid differentiation by GSK3 inactivation of GSK3 (glycogen synthase kinase 3), thus indicatingpointing to mitophagy as being a regulatory mechanism for theAML progression of[ AML96 [7]]. More recently, the loss of sequestosome 1 ( SQSTM1 ), also known as p62, a selective autophagy receptor crucial for the development and progression of AML in vivo, induces the accumulation of damaged mitochondria and mitochondrial superoxide, thusereby compromising thecell survival of. leukemia cells. Thenus, the loss of SQSTM1 impairs leukemia progression in AML mouse models of AML, underlyscoring the role of mitophagy in theLSC survival. of97 LSCs [8]]. AColltogetherectively, these studies demonstrate that enhancincreased autophagic activity of LSCs is required for malignant progression into AML.
However, in contrast to the autophagy activation of autophagy observed in AML, a loss of autophagy in healthy HSCs triggers the expansion of a population of progenitor cells in the bone marrow, giving rise toresulting in severe and invasive myeloproliferation, such as in human AML [ 53 [9]] . This apparent paradox cmany be explained by the distinct roles that autophagy canmay play during AML progression, which may be different at various stages of leukemogenesis [10][11][ 98 , 99]. Autophagy in normal HSCs may prevent the onset of cancer, as a tumor suppressive mechanism. Indeed, autophagy removes damaged organelles, such as mitochondria, and protects hematopoietic cells from genomic instability and inflammation, thus preventing the onset of leukemia. ParNoticularably, increased DNA damage, high ROS levels, aneuploidy, and an aberrant accumulation of p62/SQSTM1 have been correlated with an impaired autophagicy process, indicating a key role of autophagy in preventing tumorhe initiation [12]of tumor [100 ] . Conversely, in established cancer, autophagy may function as a favorable pathway that promotes tumor survival and tumor growth, by helping tumor cells to escape metabolic stress and death stimuli.
Some studies have also shown that the activation of the autophagic flowux only plays a role only in the initiation of AML, with a transformation from HSCs to LSCs, and, therefore, after this phasstage, autophagy is not required for disease maintenance [13][101 ] . SAML studies in MLL-AF9 AML, the most common alteration in childhood AML, indicatepoint to ATG5 or ATG7 as necessaryrequired for the onset of AML, but once the leukemic condition is established, autophagy is not required for LSC function LSC in vivo [13][14][101 , 102 ] . However, in a different AML MLL-ENL AML mouse model, Atg5 or Atg7 knockout reduced the number of functional LSCs, increased mitochondrial activation and ROS lthevels in these cells, and prolonged the survival of leukemic mice [15]. ]. In this context, during the process of leukemogenesis process, histone methylation can regulate coreentral autophagyic effectors and upstream autophagic regulators such as ATG 5 and ATG7 to influence directly affect the level of autophagy level[104 indirectly] [16]. Together, these studies suggest a highly complex and context-dependent role for autophagy in leukemic transformation with respect to the maintenance properties of LSCs in AML.
The dual role of autophagy in AML, as a promoter or suppressor of cancer in AML, is still a matter of debate. Studies have shdemownnstrated that autophagy can act as a pro- or anti-proliferative mechanism depending on the lineage and the molecular genotypic context of the disease, reflecting the degree of heterogeneity of AML [17][105 ] .
2. Regulation of Aautophagy Ggenes in Acute Myeloid Leukemia CML cells
Numerous studies have shdemownnstrated that increased autophagy in AML cells confers protection from chemotherapeuticy treatment and promotes AML cell survival.
Increased ATG7-mediated autophagy has been associated with poor clinical outcomes and a short duration of remission in AML patients [ [18]106 ]. More recently, some proteins involved in leukemica cell survival, and overexpressed in AML, have been related to ATG overexpression, underlying the interplay between autophagy and protein overexpression that promotinges leukemica cell survival [8] [19].]. Hu et al. havde shownmonstrated that a high elevated expression of SIRT1 (Sirtuin 1), a key player in mitochondrial biogenesis and autophagy-related protein, is associated with high CXCR4elevated expression of CXCR4, a negative prognostic marker in AML, and with other other proteins related to autophagy-related proteins s such as ATG5 and LC3 in primary human AML samples, indicating a potential role of the SDF-1α-CXCR4 signaling pathway in autophagythe induction of autophagy in AML cells, which further promotes their survival under stress [107 ] [20].
The transient receptor potential melastatin receptor 2 (TRPM2) ion channel, involved in maintainingthe maintenance of cell survival followingafter oxidant injurytive damage, is highly expressed in AML [21].[ By108 performing]. By performing TRPM2- depletion , Chen SJ et al. [21][ 108 ] havde shownmonstrated that the levels of ULK1, Atg7, and Atg5 protein levels are decreased in TRPM2-- depleted cells , leading to autophagy inhibition of autophagy. Importantly, theTRPM2 depletion of TRPM2 in AML inhibits leukemia proliferation and increases the doxorubicin sensitivity of AML cells [21][ 108 ].
Functional studies in normal CD34+ CB cells indicated that the inhibition of VMP1 expression reduced autophagic flux, with decreased hematopoietic stem and progenitor cell (HSPC) expansion, delayed differentiation, increased apoptosis, and impaired cellllular function and in vivo engraftment in vivo. Similar results were observed in leukemic cell lines and primary AML CD34+ AML cells. Furthermore, ultrastructural analysis indicated that leukemic cells overexpressing VMP1 have a reduced number of mitochondrial structures, and the number of lysosomal- degradingation structures hais increased. Overexpression of VMP1 (vacuole membrane protein-1) overexpression inincreased autophagic flux and improved mitochondrial quality,109 which coincided with an increased threshold for venetoclax-induced loss of mitochondrial outer membrane permeabilization (MOMP) and apoptosis in leukemia cells [22]].
Heterozygous deletions, missense mutations, or changes in thecopy number of copies of key autophagy genes have been found with a high frequency in AML patients, especiallyparticularly in AML patients with complex karyotypes [5 , [15][23]103 ] . In particular, a heterozygous chromosomal loss of 5q, 16q, or 17p correlates with regions encoding the autophagyic genes ATG10 and ATG12 , GABARAPL2 , and MAP1LC3B, or GABARAP , respectively [15][ 103 ], and severmalny others autophagyic genes have a low level of expression in hthe Human AML blasts, a decreasereduced autophagic flowux, and high levels of ROS [ [15]103]. In addiFurtion, ahermore, one study suggested that key autophagy genes such as ULK1 , ATG3 , ATG4D, and ATG5 were significantly downregulated in primary AML cells compared to normal granulocytes [ [24]110 ].
3. Autophagic Bbiomarkers
Significant progress has recently been made to identify specific autophagy-related genes for the prediction of clinical outcomes in AML. Along with the Along with the previously described ATG genes previously described, several microRNAs implicated in leukemogenesis and chemoresistance have been also also been involved in theautophagy activation of autophagy, and mayand can be used as biomarkers [25][ 111 ]. In pNotarticularbly, miR-17-5p overexpression in leukemia promotes AML proliferation by inhibiting autophagy through BECN1 targeting [26][27][28][ 112 , 113 , 114]. Ganesan et al. demonstrated that stromal cells downregulate miR-23a-5p levels in leukemica cells, leading to the upregulation of protective autophagy in these cells, thereby increasing their resistance to chemotherapy toxicity. [29]115 ]. MiR-143 oOverexpression was of MiR-143 has been shown to enhance the sensitivity of AML cells to the cytotoxicity of cytarabine (Ara-C) treatment by inhibiting autophagy through targeting ATG7 and ATG2B targeting[116 ] [30]. An overexpression of miR-15a-5p is involved in the chemoresistance of AML patients, through the autophagy-related genes ATG9A , ATG14 , GABARAPL1, and SMPD1which targeting AML cells [ [31]117 ].
Recent advances in bioinformatics have yielded an autophagy-related signature that cman help toy help predict overall survival (OS) and/or the clinical outcomes of AML patients with AML. Several studies have shown that the progression of AML depends on the autophagy-agene signature associated gene signaturewith autophagy [ 118 [32]]. A recent bioinformatics study hacons builttructed a model containing 10 autophagy-related genes to predict the survival of AML patients, showdemonstrating that groups at high risk of AML have an increasedAML risk have higher expression of immune checkpoint genes and a higher percentage of CD4 T and NK cells [33][119] .]. In addiFurtionhermore, this research study was able to predict OS in AML through the signature of 10 genes, indicapointing to this model as an effective prognostic predictor for AML patients, useful to guide patient stratification for immunotherapies and drugmedications [33][119] . . The LASSO Cox bioinformatics study LASSO Cox regression thatstudy which identified a critical risk signature for AML, consisting of the autophagyic genes BAG3 , CALCOCO2 , CAMKK2 , CANX , DAPK1 , P4HB , TSC2, and ULK1 , had excellent predictive power for AML prognosis [34][ 120]. Notably, the immunosuppressive cytokines were found to be significantly increased in the tumor microenvironment of patients with a high- risk of AML, predicted on the basis of the autophagy-related signature of these patients [34][120 ] . However, the prognostic value of the ATG signature in the clinical setting is still debated. Therefore, the roles of the ATG signature and autophagy in the pathogenesis of AML should be further investigated.
Furthermore, an interesting study indicated that an autophagy-related lncRNA signature containing six lncRNAs ( HYMAI , MIR155HG , MGC12916 , DIRC3 , C1orf220, and HCP5 ) may have an important prognostic value [35][ 121 ]. A recent study poindicated ted to four autophagy-associated lncRNAs ( MIR133A1HG , AL359715.1 , MIRLET7BHG , and AL356752.1 ) as a signatures to potentially use be used as a biomarker to predict the survival of AML patients [ 122 [36]].
Collectively, these data indicate that the role of autophagy in tumor development clearly depends on the type of AML and stage of tumor development. Furthermore, autophagy may provide cancer cells with a survival strategy, suggesting a therapeutic use for autophagy inhibition. On the other hand, autophagy can induce cell death, pointing to autophagy activation as a novel strategy in cancer therapy. Therefore, it is necessary to determine the role played by autophagy in the molecular subtypes of AML, or the degree of tumor development, to verify whether its modulation could lead to benefits for the treated patient.
4. Autophagy and Ggenetic Aalterations in Acute Myeloid MLeukemia
The AML phenotype results from multiple molecular, genetic, and epigenetic alterations thaffectingt affect the differentiation, proliferation, and apoptosis of myeloid progenitors. The World Health Organization has classified AMLs according tobased on the presence of particular genetic alterations, frequentlyoften originating from chromosomal translocations or other genome rearrangements such as t(8;21), t(15;17), inv (16), inv(3) (3) , t(6;9), t(9;11) or t(11;19), or mutations in receptor kinases, in key signaling mediators, proto-oncogenes, or epigenetic enzymes, e.g.for example, mutations in FLT3 (FMS- - like tyrosine kinase 3), TP53 , c-KIT or IDH1/2 , NPM1 (nucleophosmin 1), and CCAAT enhancer- binding protein ( CEBPA ) [ 1 [37][38][39], 2 , 123 ]. These mutations in AMLs have an impact on the choice of the most suitable therapy.
The association between autophagy and recurrent genetic alterations has been described in several AML studies in AML, but needs further investigation [ [40][41]124 , 125 ]. Here, rwesearchers summarize and update the recent advances that have highlighted the link between autophagy and fusion genes and recurrent oncogenic mutations in AML and the involvement of autophagy in chemotherapy treatment ( Figure 1 ).
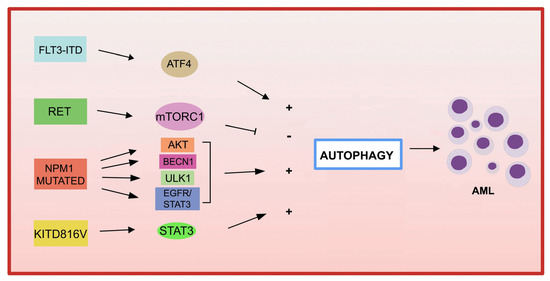
Figure 1. Gene mutations in AML and autophagy. Several recurrent genetic abnormalities in AML are involved in the deregulation of autophagy, leading to leukemia progression.
4.1. Fusion Genes in Acute Myeloid Leukemia and Autophagy
4.1. Fusion genes in AML and autophagy
Most cases of APL are caused by a de novo translocat ion t(15;17) (q22; q21) translocation, which (q22;q21), resultsing in the fusion of the RARA gene with the PML gene [ 126 , 127 [42][43]]. APL cells that have a lower expression of autophagy-related genes than normal cells have a reduced autophagic activity. By using differentiating agents, such as all-Using differentiating agents, such as all- trans- retinoid acid (ATRA) and arsenic trioxide (ATO) currently used in clinical settings, the expression level of autophagy-related genes increases, thus restoring autophagy in APL cells [ [44]128]. Both agents can activate ETosis, a type of cell death mediated by the release of neutrophil extracellular traps (ETs). In additionFurthermore, mTOR-dependent autophagic action is required for ATO-induced NETosis in APL cells [ [45]129 ]. Of note, rapamycin, the inhibitor of the mTOR pathway, synergizes with ATO in the eradication of leukemia-initiating cells (LIC) through the activation of NETosis in both APL cells and an in vivo APL model [45][129 ] .
M
Translocations of the mixed lineage leukemia ( MLL ) gene translocations 11q23 wegene are observed in approximately 80% of pediatric AMLs. In these, the MLL gene can, by genomic translocation, be fused with >60 different fusion partners [46][ 130 ]. Treatment with the RAS oncogene inhibitor, tipifamib, leads to the inhibition of AML withby the t(6;11) translocation by inducing both apoptosis and autophagy [47][ 131 ]. Another study demonstrated that ATG5 participates in the development of MLL-AF9-- driven leukemia , but not in AML-sensitive chemotherapy mice expressing MLL-AF9 [ 132 [48]].
Acute myeloid leukemia with core binding factor ( CBF-AML ) is characterized by the presence of t(8;21) (q22; q22), or inv (16) (p13q22)/t(16;16), which leadsleading to the formation of RUNX1/RUNX1T1 (AML1/ETO) and CBFbeta-MYH11 , respectively [49].[ The133 activation]. of ULK1-mediated autophagy may control and delay ULK1-mediated activation of autophagy can control and delay AML1-ETO9a- -gudridedven leukemogenesis in an AML CASPASE-3 knockout mouse model [50][ 134], suggesting that CASPASE-3 is an important regulator of autophagy in AML. The results of these studies highlight the different roles of autophagy in the initiation, progression, and chemotherapeuticy responses in AML cells, depending on the different type of aberrant oncoprotein.
4.2. Gene mutations in AML and autophagy
4.2. Gene Mutations in Acute Myeloid Leukemia and Autophagy
-
FLT3
FLT3
Among the most common genetic alterations in AML, mutation of the tyrosine kinase 3 ( FLT3 ) gene mutation ooccurs in approximately 30% of AML cases.
The most frequent aberrations affecting the FLT3 gene , associated with a poor prognosis in AML, are the internal tandem duplication ( FLT3 -ITD) in the juxtamembrane domain, and point mutations, involving the tyrosine kinase domain of FLT3 ( FLT3 -TKD) [ [51]135 ]. Expression of FLT3 -ITD expression inincreases basal autophagy in AML cells through a mechanism involving the transcription factor ATF4 (activating transcription factor 4) [52][ 136 ]. In addiFurtionhermore, the inhibition of autophagy in FLT3 cells-TKD cells, which are resistant to the FLT3 inhibitor quizartinib (AC220), also inhibits proliferation both in vitro and in vivo [ [52]136 ]. More recently, the acquired D835Y mutation induced resistance to the FLT3 inhibitor sorafenib, and activated autophagy in FLT3 -ITD-positive cell lines. By inhibiting autophagy, the authors were able to overcome resorafenib resistance to sorafenib iin FLT3 -ITD-positive AML, improving its efficacy [53][ 137 ]. Recently, a study demonshowtrated that the inhibition of autophagy reduces the repopulation potential of FLT3-ITD AML LSCs associated with mitochondrial storaccumulationge-associated [54]FLT3 -ITD AML LSCs [ 138]. In addiFurtionhermore, the authors showdemonstrated that autophagy inhibition improvenhances p53 activity and increases theenhances TKI-mediated inhibition of AML progenitors [54][ 138 ].
Autophagy not only contributes to the downstream proliferation of the FLT3-ITD receptor, but may also be involved in the degradation of the mutated receptor degradation. In fact. Indeed, in one study, the frequent activation of the RET receptor tyrosine kinase RET was observed in several AML different subtypes of AML [ [55]139 ]. RET mediates autophagyhe suppression of autophagy in an mTORC1-dependent manner, leading to the stabilization of the mutant FLT3 receptor. The geGenetic or pharmacological inhibition of RET decreasereduced the growth of FLT3-dependent AML cells, with the upregulation of autophagy and FLT3 depletion of FLT3 [ [55]139]. These results suggest that restoratingon of autophagy in FLT3-dependent AML may result in the degradation of mutated FLT3,nt FLT3 and thereforeus may represent an interestingattractive therapeutic approach. It has also been shown that the inhinhibition of the FLT3-ITD protein leads to anhas also been shown to lead to increase ind ceramide synthesis and mediates ceramide-dependent mitophagy, leading to AML cell death [56][57][140 , 141 ] .
-
KIT
Mutations in KIT
KIT mutations are associated with an increased leukemia cell proliferation of leukemic cells and an increased risk of AML recurrence [58][59][ 142 , 143 ]. A recent study reported that the KIT D816V mutation in AML cells increases basal autophagy, stimulating AML cell proliferation and survival via STAT3 signaling [60][ 144 ]. A different point mutation in c- KIT (N822K T > A) constitutively activates this receptor, making AML cells highly sensitive to sunitinib (a tyrosine kinase inhibitor), resulting in AML cell death through the activation of both apoptosis and autophagyic processes [ 145 [61]].
NPM1
-
NPM1
Mutations in NPM1 (nucleophosmin 1) are the most frequent genetic alterations in adult AML, responsible for the aberrant localization of the NPM1 protein in the cytoplasm [62].[ Increased146 autophagic]. activity found in The increased autophagic activity found in NPM1 -mutated AML cells is involved in leukemic cell survival [ [63]147 ]. The MNPM1 mutant NPM1 can also interact with the tumor suppressor protein PML (leukemia pro-myelocytic protein), leading to PMLthe delocalization and stabilization that of PML which, in turn, can activate autophagy via AKT signaling [63][147 ] . In another study, it was shown that in AML patients carrying the mutant NPM1 , the glycolytic enzyme PKM2 (pyruvate kinase M2) induced autophagy viathrough phosphorylation of the autophagic protein Beclin 1, contributing to cell survival [ [64]148 ]. Finally, the NPM1 mutant protein can also interact with the autophagic protein ULK1, stimulating the TRAF6-dependent ubiquitination of ULK1 via miR-146, thereby maintaining ULK1 stability and functionality and promoting autophagic cell survival [65][149 ] . Furthermore, it was observed that thhe expression of RASGRP3, a protein associated with tumor progression, is was observed to be upregulated in AML patients with AML with NPM1 mutation compared to AML patients with AML without mutant NPM1. . The authors demonstrated that NPM1 -mut blocks the degradation of the RASGRP3 protein through binding to the E3 ubiquitin ligase E3 MID1 protein, leading to RASGRP3 overexpression of RASGRP3, as well as promoting the downstream activation of EGFR-STAT3, which in turn promoteds proliferation and autophagy in AML cells [66][150 ] .
P53
-
P53
Alterations inof the tumor suppressor gene TP53 are found in abpprout 5–ximately 5-15% of AML cases, and, and frequently, in older patients [67][68].[ It151 has, been152 proposed]. thatIt has been proposed that the role of autophagy in the development of AML may be determined by TP53 the role of autophagy in the development of AML can be determined by the status of TP53. For wild-type AML TP53 AML, researchers have showndemonstrated that pharmacological ablockade of autophagy blockade achieves therapeutic benefits, whileereas AMLs harboring TP53 mutations dofail notto respond to theautophagy inhibition of autophagy by hydroxychloroquine (HCQ) [69][70][ 153 , 154]. The use of autophagic inhibitors may be a potential therapeutic strategy to use, particularly for the treatment of TP53 wild-type TP53 AML. For AML with TP53 mutations , autophagic pathways may be a therapeutic option to usebe used for the elimination of the TP53 T mutant T TP53.
Another study demonstrated that stimaulation of macroautophagy stimulation by 17-AAG, an HSP90 inhibitor, causesresults in the degradation of TP53 R248Q in AML cells and also enhances the transcription of autophagy-associated genes [71][155 ] . In addiFurtionhermore, accumulateding evidence indicates that TP53 activated by a variety of cellular stresses can trigger autophagy through the transactivation of pro-autophagyic genes, including DRAM1DRAM1 ( DNA damage regulated (autophagicy modulator regulated by DNA damage 1), SESN1 (sestrin 1), and ( sestrin SESN2 ) [ [71][72][73][74]155 , 156 , 157 , 158 ].
A recent study highlighted the role of autophagy in AML cells, in the context of p53-mediated apoptosis, which is associated with increased cytotoxicity to treatment with MDM2 inhibitors and Ara-C when miR-10a is inhibited [75][159 ] . The antileukemic strategy based on the use of MDM2/X inhibitors in wild-type p53 tumors to restore the normal and active conformation of p53, MDM2, and MDMX has not been extensively tested [160 [76]] . Thuserefore, the use of a combination of treatments, including MDM2 inhibitors with autophagic modulators, maycould be a newovel strategy to improve the treatment of wild-type p53 AML .
Pharmacological treatments that modulate autophagy in AML patients carrying p53 mutations participate in the degradation of aberrant p53 proteins. The pPoint mutation of TP53 at the amino acid residue R428 (R248Q), with gain-of-function activity, gives rise to malignant activity in lung cancer cells [ 161 [77]] and a loss of tumor suppressor function in AML [78][ 162 ].
Interestingly, the treatment with the Hsp90 inhibitor (17-AAG) results in the activation of chaperone-mediated autophagy, which induces the degradation of the aberrant protein p53R248Q in AML cells. In particularNotably, under conditions of metabolic stress, 17-AAG induces the interaction between p53R248Q and the chaperone protein Hsc70, triggering chaperone-mediated autophagy to degrade p53R248Q [ [71]155 ]. These data open new opportunities for future studies that maycould elucidate the functional involvement of different types of autophagy and their connection with the molecular mechanisms tofor improveing anticancer therapies against AML harboring the different TP53 variants.
IDH1/2 (isocitrate dehydrogenase)
.
-
IDH1/2 (isocitrate dehydrogenase)
Recent advances in bioinformatics have enabled the identification of several epigenetic mutations affecting AML, including IDH1/2 , Tet methylcytosine dioxygenase 2 ( TET2 ), DNA methyltransferase 3A ( DNMT3A ), and ASXL1 , all of which are associated with the pathogenesis of AML [ 163 , [79][80][81]164 , 165]. IDH proteins are isocitrate dehydrogenases, implicated in various biological processes, such as energy metabolism, histone demethylation, DNA modification, and adaptation to hypoxia. Further studies are needed to investigate innovative therapies based on targeted autophagy in combination with DNA hypomethylation to treat AMLs harboring certain types of epigenetic alterations.
DNMT3A
Mutations in the
-
DNMT3A
Mutations in the DNMT3A gene , an enzyme involved in the methylation of CpG dinucleotides methylation, are present in 20–-23% of adult patients with de novo AML [81][ 165 ]. Several studies have shown that the treatment of AML patients with DNA methyltransferase -inhibiting agents, such as azacitidine (5-aza-2′-deoxycytidine), induces autophagic activity in AML leukemia cells [82][166 ] . A study performed oin a DNMT3A R878H conditional knock-in mouse model, used to predict the specific long non-coding RNAs (lncRNAs) regulated by DNMT3A mutations in AML, first identified 23 lncRNAsdifferentially expressed lncRNAs, then the downstream target genes regulated by these lncRNAs, including ATP6V1A , a critical autophagy-related gene, the overexpression of which is associated with poor prognosis in AML [83].[ However,167 there]. is still little evidence of a direct involvement of However, there is still little evidence for a direct involvement of DNMT3A gene mutations with autophagic activity in AML.
Further studies are needed to understand the functional significance of autophagy associated with different genetic mutations in AML cells.