Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Joseph AMJL Janssen and Version 3 by Joseph AMJL Janssen.
The metabolic syndrome is a cluster of overlapping conditions resulting in an increased incidence of type 2 diabetes, cardiovascular disease, and cancer. Prevalence of the metabolic syndrome in the Western world has reached epidemic proportions and this is likely due to alterations in diet and the environment as well as decreased physical activity.
- hyperinsulinemia
- IGF-I
- IGFBP-1
- insulin resistance
- impaired glucose tolerance
- diabetes
- metabolic syndrome
- nutrition
- (im)balance of the insulin-GH-IGF-I axis
1. Introduction
The metabolic syndrome (previously also called syndrome X and the insulin resistance syndrome) is characterized by a group of metabolic disturbances which are associated with an enhanced risk of type 2 diabetes, cardiovascular diseases, certain types of cancer, and mortality. Over the last fifty years, the metabolic syndrome in the Western world has reached epidemic proportions. Since the human genome has been virtually unchanged since 1950, the increased prevalence of the metabolic syndrome can be attributed to important environmental changes interacting with genes [1].
The “modern” Western diet is characterized by a high content of saturated fats, sugar, corn-derived fructose syrup, proteins (derived from fatty domesticated and processed meats), refined grains, low fiber content, salt, and reduced consumption of fruits and vegetables [2]. The “modern” Western diet and lifestyle may have played an important etiological role in the pathogenesis of the metabolic syndrome and its consequences.
2. The Insulin–IGF-I System
The insulin–IGF system is formed by insulin, two insulin-like growth factors (IGF-I and IGF-II), and six cell-membrane signal-transducing receptors (insulin receptor-A (IR-A), insulin receptor-B (IR-B), insulin-like growth factor-I receptor (IGF-IR) and their respective hybrids): the IRs and IGF-IR can be found at the cell surface as homodimers composed of two identical alpha/beta monomers, or as heterodimers composed of two different receptor monomers (aka hybrids) [3][4][5][3,4,5]. The system further contains the IGF-II scavenger transmembrane protein previously known as insulin-like growth factor “receptor”-II “(IGF2R), and six IGF-binding proteins (IGFBP-1-6), several IGFBP-related proteins and IGFBP proteases, which serve to tightly regulate IGF-I and IGF-II levels/bioavailability in circulation and at the cell surface [3][4][5][6][7][8][3,4,5,6,7,8]. Insulin and IGF-I show high structural similarities. It has been postulated that IGF-I emerged at a very early stage in vertebrate evolution from an ancestral insulin-type gene [9]. This ancestral insulin-like gene was duplicated to form insulin and IGF-I in early (agnathan) vertebrates. Further gene duplications resulted in distinct IGF-I and IGF-II genes, which were first found during evolution in gnathosomes (jawed vertebrates) [10]. In vertebrates, insulin-like peptides predominantly functioned as growth factors promoting tissue growth and development. However, in vertebrates, this growth-promoting function has been subsumed by IGF-I and IGF-II, while insulin has acquired this new function and is primarily involved in metabolism. It has further been demonstrated that, in vertebrates, nutritional status is the key regulator of the activities of insulin and IGFs [11]. Consequently, insulin and IGFs regulate metabolism, growth and development in vertebrates in response to nutritional availability. IGF-I displays approximately 50% structural homology with proinsulin [12]. In humans, insulin and IGF-I are transported in plasma and delivered to cell membrane receptors [13]. In contrast to insulin, IGF-I is mainly present in the plasma bound to six different binding proteins, the so-called IGFBPs. The IGFBPs act as transport proteins and control efflux from circulating IGF-I and IGF-II [13]. IGFBPs modulate interactions between the IGFs and the IGF-IR, thereby indirectly controlling the biological actions of IGF-I and IGF-II [13]. Because of the high affinity of IGF-I for IGFBPs, <1% of circulating plasma IGF-I is in a free form, whereas in contrast, insulin is largely in a free form [14]. Free IGF-I may be the major biologically active hormonal form of IGF-I [15]. When evaluating IGF-I bioactivity in health and disease, it is therefore recommended also to measure circulating free IGF-I levels [15].3. The Role of Insulin and IGF-I in Metabolism
Insulin and IGF-I are both complementary and involved in metabolism, growth, and in the control of essential cellular and physiological processes such as proliferation, differentiation, and body growth [16]. Although insulin is, in many respects, structurally similar to IGF-I, insulin binds with high affinity to the insulin receptor, whereas it has very low affinity for the IGF-I receptor, while the converse is true for IGF-I [17]. Binding of IGF-I to the IGF-I receptor primarily results in stimulation of proliferation and differentiation of cells [17]. Nevertheless, IGF-I also stimulates insulin-like actions (glucose and amino acid uptake, mRNA, and protein synthesis) in cells and enhances insulin sensitivity [14][18][14,18]. Most of the ability of IGF-I to enhance insulin sensitivity is mediated (indirectly) through suppression of GH, a well-known insulin antagonist [14]. In addition, IR–IGF-IR hybrids behave like IGF-IRs but show differential IGF-I vs. IGF-II stimulated activity based upon the involved IR isoforms [19][20][19,20]. Healthy adult liver cells show a very low expression of the IGF-IR, and the IGF-IR expression in fat cells of the adipocyte tissue is also low and tends to decrease when differentiating from pre-adipocytes to mature adipocytes [21][22][21,22]. Therefore, these latter tissues, while being extremely insulin responsive, are relatively refractory to exposure to IGF-I [14]. A complex inter-relationship exists between IGF-I, insulin, and GH [23]. Hepatic IGF-I production is stimulated by both GH and insulin, while IGF-I feeds back to suppress GH and insulin secretions [23] (see further below).4. Reaven and the Metabolic Syndrome
Reaven previously suggested that the more insulin sensitive an individual, the better off (s)he is [24]. In an insulin-sensitive individual, the beta-cell is not stressed, and glucose tolerance can be maintained at low plasma insulin levels [24]. However, he also suggested that resistance to insulin-stimulated glucose uptake is present in more than 80% of individuals with impaired glucose tolerance or type 2 diabetes, and in about 25% of nonobese individuals with normal glucose tolerance [24]. Insulin resistance, by itself, is not sufficient to produce diabetes [24]. If the pancreatic beta-cells are able to overcome insulin resistance by increasing their insulin secretory response, frank type 2 diabetes will not occur. Thus, hyperinsulinemia may prevent frank diabetes in insulin-resistant individuals. However, according to Reaven, the compensatory hyperinsulinemia to overcome insulin resistance has its own price (independent from the development of diabetes) and may produce unwanted effects [24]. In 1988, Reaven hypothesized that insulin resistance and hyperinsulinemia induce the metabolic syndrome: insulin resistance and hyperinsulinemia provide a common mechanism for the development of glucose intolerance, elevated circulating triglycerides, decreased HDL-cholesterol and hypertension [24]. In the view of Reaven, glucose intolerance, elevated circulating triglycerides, decreased HDL-cholesterol, and hypertension are more likely to occur together than separately, and when they cluster, they are highly likely to be the manifestations of resistance to insulin-stimulated glucose uptake and (compensatory) hyperinsulinemia [25]. Reaven further proposed that the metabolic syndrome plays both an important role in the etiology and clinical course of three major related conditions: hypertension, coronary artery disease and type 2 diabetes [24]. The prevalence of the metabolic syndrome varies from 8% to 67% among different populations, and it has been suggested that the variety in prevalence of the metabolic syndrome and its individual components may be related to differences in genetic background, diet, levels of physical activity, population age and sex structure, levels of over- and undernutrition, body habitus, and definitions of the metabolic syndrome [26]. Insulin resistance and hyperinsulinemia appear to be central in the development of the metabolic syndrome [27][28][27,28]. However, at present it is still unclear whether there is a unifying mechanism which causes the metabolic syndrome.5. Causes of Hyperinsulinemia
In the view of Reaven (see above) and in most traditional literature, obesity is considered the main cause of insulin resistance. In the view of Reaven insulin resistance is the abnormality leading to hyperinsulinemia. In this model, hyperinsulinemia is a compensatory response to insulin resistance. However, an increasing number of (prospective) human studies suggests an alternative scenario: in this scenario, chronic hypersecretion of insulin is the primary abnormality leading to hyperinsulinemia and precedes, initiates, and causes insulin resistance. In this new scenario, hyperinsulinemia is the first event triggering insulin resistance, obesity, the metabolic syndrome, and type 2 diabetes [29][30][31][32][33][34][35][36][37][38][39][29,30,31,32,33,34,35,36,37,38,39]. In addition, in subjects with normal plasma glucose concentrations, it has been found that hyperinsulinemia per se induced insulin resistance by insulin-induced downregulation of insulin receptor signaling [40][41][40,41]. This further supports the idea that hyperinsulinemia may be a primary driver of insulin resistance [41][42][41,42]. Moreover, it has been shown that gastric bypass surgery in type 2 diabetes corrects and normalizes plasma insulin levels within some days after surgery, even though there is, at that moment, no significant weight loss (yet) and insulin resistance remains high [43]. This post-operative course clearly dissociates hyperinsulinemia from insulin resistance, further supporting the idea that hyperinsulinemia is primary, rather than a consequence occurring secondary (as a compensation) to insulin resistance [43][44][43,44]. In a well-characterized cohort of apparently healthy adults, elevated fasting insulin at baseline was found to be an independent determinant over a 5-year period for the future development of the metabolic syndrome [45]. In addition, prospective evidence shows that—independently of obesity and body weight—hyperinsulinemia is related to the development of dyslipidemia and hypertension, suggesting that hyperinsulinemia precedes these disorders in the etiologic pathway [46][47][48][46,47,48]. Thus, hyperinsulinemia may indeed be an early and central feature of the cardiovascular risk of subjects with the metabolic syndrome [45]. Further support of a pathogenic role of hyperinsulinemia in the development of the metabolic syndrome was demonstrated in an animal model by Jeanrenaud et al. They showed that short-term hyperinsulinemia is a pathological driving force, which produces incipient obesity by overstimulating white adipose tissue and liver metabolic activity while concomitantly producing incipient muscle insulin resistance [49]. In her Banting Lecture in 2011, Barbara Corkey proposed a model in which excessive beta-cell insulin secretory responses, possibly induced by consumption of the “modern” Western diet and over-nutrition, superimposed on a susceptible genetic background and metabolic programming, may be a major cause of hyperinsulinemia, insulin resistance, obesity, and type 2 diabetes [42]. As Corkey pointed out, many aspects of the Western diet may potentially promote hyperinsulinemia: excess nutrient ingestion, artificial sweeteners, mono-oleoylglycerol, the macronutrient ratio in the food, the characteristics of the carbohydrates, proteins and fat of the food, and insufficient dietary fiber intake [50]. In the model of Barbara Corkey, hyperinsulinemia is the dominant driver of insulin resistance. In her model, insulin resistance is an adaptive response protecting muscles from chronic hyperinsulinemia-mediated nutrient excess and intracellular hyperglycemia [42][51][42,51]. Increased activity of incretin hormones may also contribute to the development of hyperinsulinemia. Gastric inhibitory polypeptide (GIP) is an incretin hormone secreted by the gut leading to enhanced pancreatic insulin secretion after food ingestion [52]. A significant elevation of fasting GIP was found in healthy young men after five days of ingesting a Westernized diet (i.e., a diet high in fat and calories: containing 50% extra energy, where 60% of the energy came from fat (HFHC diet) [37]. In this latter study, insulin secretion was increased and preceded the development of peripheral insulin resistance, mitochondrial dysfunction, and obesity in response to overfeeding, suggesting a direct and causal role for the Westernized diet, GIP, and hyperinsulinemia in the development of peripheral insulin resistance and obesity [37]. Pancreatic insulin secretion and (hepatic) insulin clearance play distinct roles in peripheral plasma insulin concentrations. Emerging evidence suggests that not only pancreatic insulin secretion, but also lower hepatic insulin clearance, may contribute to hyperinsulinemia (=increased peripheral insulin levels). A wide variation in hepatic as well as extrahepatic insulin clearance in human subjects has been reported [53]. Hepatic insulin clearance is (at least partly) a heritable trait; several genetic variants that are involved in regulating insulin clearance have been identified in Mexican Americans, an ethnic group with a high prevalence of the metabolic syndrome and diabetes [53]. After an overnight fast, hepatic first-pass insulin extraction in Afro-Americans (AAs) was found to be only one third compared to European Americans (EA) [54]. Bergman et al. suggested the following course of events in the development of type 2 diabetes: low(ered) hepatic insulin clearance causes peripheral hyperinsulinemia, which in turn exacerbates insulin resistance [54]. Consequently, insulin resistance will stress pancreatic beta-cells, and this may finally result in their ultimate failure and onset of frank type 2 diabetes [54]. Therefore, Bergman et al. hypothesized that low(ered) hepatic insulin clearance and peripheral hyperinsulinemia can be a primary cause of type 2 diabetes in high-risk individuals [54]. In their view, development of insulin resistance in muscles and fat cells is caused by of an overexposure of the (post-hepatic) peripheral tissues to endogenous insulin [54]. As a direct consequence of the peripheral hyperinsulinemia, peripheral insulin resistance develops to dampen hyperinsulinemia-mediated stimulating effects in muscles and fat cells [54]. Reduced hepatic insulin clearance has been reported in healthy lean subjects after only three days on a diet containing high carbohydrate (80E%), low fat (9E%), and a 75% increase in daily energy calories [55]. This suggests that a high-carbohydrate, high-calorie diet may quickly and directly change hepatic insulin clearance [55]. By contrast, energy restriction induced by Roux-en-Y gastric bypass (RYGB) surgery of obese subjects with normal glucose tolerance, and obese patients with preoperatively type 2 diabetes, increased hepatic insulin clearance (=normalized) significantly within one week after operation [56]. All these studies suggest that hepatic insulin clearance is a dynamic process which can be modified (within a few days) by diets with altered energy and, particularly, carbohydrate intake. In addition, all these studies point to an important role of reduced hepatic insulin clearance as the potential culprit in the development of (peripheral) hyperinsulinemia [57].6. The Effects of Hyperinsulinemia on the Balance of the Insulin–GH–IGF-I Axis
The insulin–growth hormone–IGF-I (insulin–GH–IGF-I) axis plays a pivotal role in metabolism. Nutrients stimulate the activity of the insulin–GH–IGF-I axis, and insulin, GH and IGF-I secretion are all nutritionally regulated [14][58][14,58]. Insulin secretion into the portal venous system is required for normal liver GH responsiveness and hepatic IGF-I synthesis and bioavailability [59]. Insulin is essential for growth hormone receptor expression in the liver and growth hormone-mediated hepatic IGF-I production [60]. There is also evidence that insulin may directly stimulate hepatic IGF-I expression [61]. Insulin (produced in the pancreas) and GH (produced in the pituitary gland) both stimulate IGF-I production in the liver, while, after secretion, IGF-I feeds back to suppress both insulin and GH secretion [60]. In healthy individuals, the insulin–GH–IGF-I axis is in balance: insulin and GH stimulate hepatic IGF-I production, whereas IGF-I secreted by the liver feeds back to suppress both insulin and GH [62][63][62,63] (Figure 1A). The typical modern Western diet can disturb this balance. Due to its continuous food intake, energy surplus, high content of sugars, corn-derived fructose syrup, saturated fats and proteins, the modern Western diet may induce hyperinsulinemia, which in turn increases IGF-I secretion [63]. Increased IGF-I subsequently induces suppression of GH secretion to lower levels than normal [64][65][64,65]. It has been found that only a few days of overeating markedly suppressed GH secretion (before any measurable weight gain) and the accompanying hyperinsulinemia is a likely mediator of this rapid reduction in GH secretion [66]. Thus, nutritionally driven hyperinsulinemia may disturb the normal balance of the insulin–GH–IGF-I axis by shifting the insulin : GH ratio towards insulin (and IGF-I) and away from GH [63] (Figure 1B). The increased insulin : GH ratio stimulates energy storage and lipid synthesis and inhibits lipid breakdown and thereby promotes obesity by promoting higher fat accumulation and lower energy expenditure [63]. Nutritionally driven disbalance of the insulin–GH–IGF-I axis may be an important etiological factor in the development of metabolic syndrome, impaired glucose tolerance and type 2 diabetes. Intriguingly, the (imbalance of the) insulin–IGF-I system is increasingly being implicated in the development of cardiovascular disease and cancer [67][68][67,68].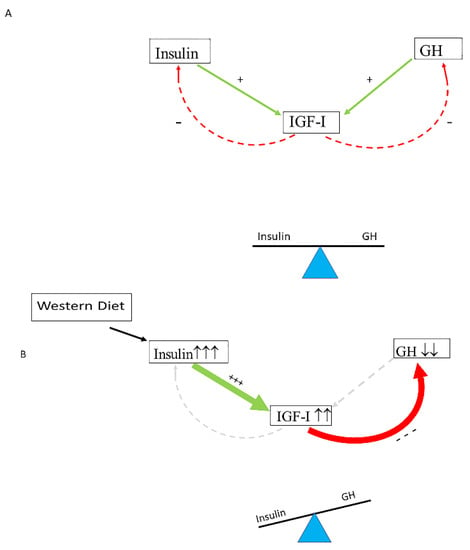
Figure 1. Nutritionally driven hyperinsulinemia can disturb the normal balance of the Insulin–GH–IGF-I axis. (A) In healthy individuals, the insulin–GH–IGF-I axis is in balance: insulin and GH stimulate hepatic IGF-I production, whereas IGF-I secreted by the liver feeds back to suppress both insulin and GH. (B) The modern Western diet may induce hyperinsulinemia, which in turn increases IGF-I secretion. Increased IGF-I levels subsequently induce suppression of GH secretion to lower levels than normal. Only a few days of overeating may markedly suppress GH secretion before any measurable weight gain, and it has been suggested that, in these circumstances, the accompanying hyperinsulinemia is a likely mediator of this rapid reduction in GH secretion. Consequently, a shift of the insulin : GH ratio towards insulin (and IGF-I) and away from GH will occur. The higher insulin : GH ratio lowers energy expenditure and induces fat accumulation, thereby promoting energy storage and lipid synthesis and hindering lipid breakdown. This will promote obesity because of higher fat accumulation and lower energy expenditure (see text for more details on how the Western diet may influence the balance of the insulin–GH axis). +/green: stimulating; −/red: inhibiting. +++ strong stimulation, --- strong inhibition, ↑↑↑ marked increase, ↓↓ moderately decrease.

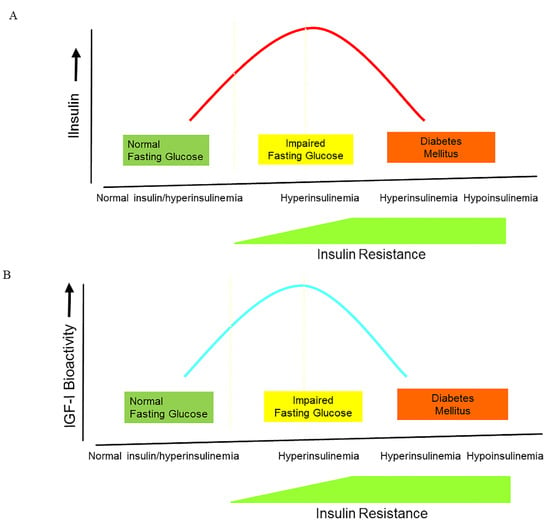
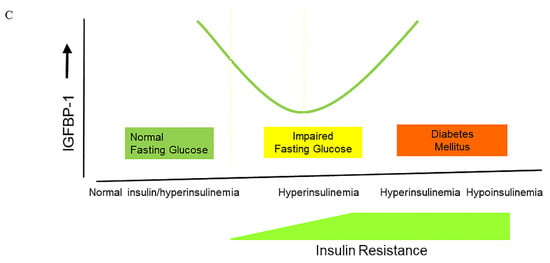

Figure 2. The changes in insulin, IGF-I bioactivity, and IGFBP-1 levels during the natural course from normoglycemia to impaired glucose intolerance and frank type 2 diabetes. (A) During the natural course from normoglycemia to frank type 2 diabetes, insulin secretion follows the pattern of an inverted U, also termed ”Starling’s curve of the pancreas”. (B) During the natural course from normoglycemia to frank type 2 diabetes, IGF-I bioactivity follows the pattern of an inverted U, suggesting that there is also a “Starling Curve” for IGF-I bioactivity. * (C) There is a U-shaped relationship between insulin and IGFBP-1 during the natural course from normoglycemia to frank type 2 diabetes **.