Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by francesco vassalli and Version 2 by Catherine Yang.
Malignant hyperthermia is a rare but life-threatening pharmacogenetic disorder triggered by exposure to specific anesthetic agents. Although this occurrence could affect virtually any patient during the perioperative time, the pediatric population is particularly vulnerable, and it has a five-fold higher incidence in children compared to adults.
- malignant hyperthermia
- pediatric anesthesia
- general anesthesia
1. Introduction
Since the beginning of the 20th century, cases of increased body temperature related to general anesthesia (GA) have been observed, and several reports referred to complications and anesthesia-related deaths, often described as “ether convulsions” [1][2][1,2].
Malignant hyperthermia (MH) is a rare but life-threatening heterogeneous pharmacogenetic disorder due to the dysfunction of skeletal muscle calcium channels, triggered by exposure to volatile halogenated anesthetics (desflurane, isoflurane, sevoflurane, halothane) and the depolarizing muscle relaxant succinylcholine, which results in abnormal contraction and a hypermetabolic state at the level of the myocyte that rapidly leads to ATP depletion, rhabdomyolysis, and heat production [3][4][5][6][3,4,5,6]. It was first described in 1962 as cases occurring in a single family by Denborough et al., when the authors reported a 21-year-old student who, when admitted to the Royal Melbourne Hospital in Australia for a leg fracture, was more concerned about receiving GA than about his fracture because 10 of his family members had died during or after GA, usually administered for minor procedures [3].
2. Epidemiology
Many physicians may believe that MH is so rare that most professionals will probably never face a case. However, there is a wide-ranging estimate of MH incidence [7]. The actual frequency of perioperative MH is challenging to estimate due to reluctance to publish adverse events, frequent misdiagnosis, and even poor adherence to pharmacovigilance registries [6][7][8][9][6,7,8,9]. Although most reported MH crises (>80%) occur in phenotypically normal children without a family history of MH-related comorbidities, we know that malignant hyperthermia susceptibility (MHS) is an autosomal dominant genetic condition that shows incomplete penetrance and affects all ethnic groups [6][7][6,7]. It is more common in males than females (2:1), with an estimated incidence of 1:10,000 in children and 1:50,000 in adults [10]. Indeed, MH mainly affects young patients, with a mean age of 18.3 years, probably due to increased penetrance in children, which, in turn, varies according to a specific genetic variant [6][10][11][12][6,10,11,12]. A higher concentration of MH-susceptible families is reported in Wisconsin and the upper Midwest in the United States [13].
3. Pathophysiology
The root cause of MH is a dysfunction of excitation–contraction coupling in skeletal muscles, leading to excessive intracellular calcium (Ca2+) release (Figure 1).
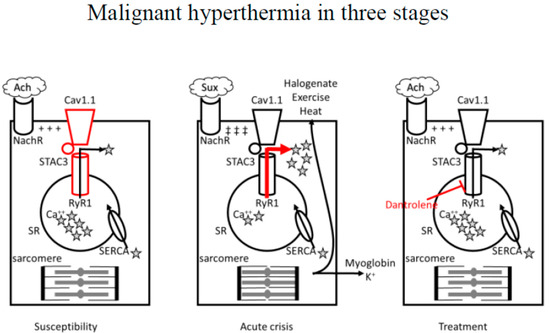
Figure 1. Role of dysfunction in the mechanism of excitation–contraction coupling in skeletal muscles primarily due to mutations in type 1 RyR1, Cav1.1, or STAC3 accessory protein in determining susceptibility to MH. Calcium release and uptake (arrows) is in equilibrium.
In susceptible patients, exposure to halogenated volatile anesthetics (halogenate), depolarizing muscle relaxant succinylcholine, exercise, or heat could trigger an acute MH crisis characterized by excessive Ca2+ release (red arrow) from the sarcoplasmic reticulum with abnormal contraction and hypermetabolic state at the level of the myocyte that rapidly leads to ATP depletion, rhabdomyolysis, hyperkaliemia, and heat production. Dantrolene sodium, a post-synaptic muscle relaxant, can effectively revert an MH crisis by inhibiting RyR1-mediated intracellular calcium release from the sarcoplasmic reticulum of skeletal muscle cells (red blunted arrow).
RyR1: ryanodine receptor. Cav1.1: L-type voltage-gated calcium channel. MH: malignant hyperthermia. Ach: acetylcholine. NachR: nicotinic acetylcholine receptor. ★Ca++: calcium. Sux: succinylcholine. SERCA: sarcoendoplasmic reticulum calcium ATPase. ATP: adenosine triphosphate. SR: sarcoplasmic reticulum. +++: end-plate potential. ‡‡‡: prolonged end-plate potential.
Under physiologic conditions, following the release of acetylcholine at the neuromuscular junction, activation of the nicotinic receptor, and depolarization of the cell membrane, the dihydropyridine receptor (DHPR, also known as L-type voltage-gated calcium channel Cav1.1) on the T-tubular membrane is activated, and after direct interaction with Type 1 ryanodine receptor (RyR1) on the sarcoplasmic reticulum (SR) membrane, Ca2+ stored in the SR is released and becomes available for stimulation of the contractile apparatus [14]. Following muscle contraction, the sarcoendoplasmic reticulum calcium ATPase (SERCA) pump acts to transport Ca2+ from the cytosol back to the SR.
In MH, the leading abnormality is due to mutations in the gene RYR1, on chromosome 19q13.1, encoding for RyR1: missense mutations alter the receptor with gain-of-function mutations, inducing increased Ca2+ release into the cytoplasm [11]. Mutations in the gene RYR1 are also associated with three congenital myopathies and an isolated case of congenital myopathy characterized on histology by cores and rods [11]. More rarely, mutations in the α1 subunit of DHPR encoded by the gene CACNA1S may be involved: by suppressing the Ca2+ voltage-gated channel’s regulatory effect on RyR1, those variants can also cause an increased Ca2+ flux through the receptor [13][14][13,14]. Finally, mutations have been identified in the STAC3 accessory protein, required to correctly locate the calcium voltage-gated receptor within the skeletal muscle channel: those variants determine an increased amount of Ca2+ released in response to caffeine (a RyR1 agonist) and increase the amount of Ca2+ stored within the SR [11][12][13][14][11,12,13,14]. However, the paucity of clinical information surrounding the MH (or MH-like) episodes noted in patients with STAC3 variants, and the lack of robust experimental evidence in in vitro contracture testing, casts some doubt on an association between STAC3 variants and MHS [11][12][13][14][15][16][17][18][11,12,13,14,15,16,17,18].
3.1. Anesthetic-Induced MH
The most well-known risk factor for MH is the use of volatile anesthetic agents, such as halothane, isoflurane, sevoflurane, desflurane, and the depolarizing skeletal muscle relaxant succinylcholine [5][6][8][5,6,8]. Halothane-induced MH seems to contribute to most MH crises; however, of all volatile anesthetics, the prevalence of MH was highest when using sevoflurane [19]. Succinylcholine administered alone is reported to trigger adverse events in approximately 15.5% of MHS patients [20]. Combining inhaled anesthetic agents and succinylcholine can significantly increase the risk of MH. Although rare, one report suggested a triggering role for amide local anesthetics (lidocaine and bupivacaine), but it has never been confirmed [21]. Considering the broad use of local anesthetics in the susceptible population without new reports and the possibility of some unrecognized interaction with volatile agents, a relationship seems to be unlikely [22].
3.2. Non-Anesthetic Induced MH
MH may occur upon exposure to other factors, even in the absence of classic anesthetic triggering agents. Increasing evidence indicates that environmental heat stroke and exertion rhabdomyolysis caused by vigorous exercise and environmental heat can induce a life-threatening hyperthermic crisis in susceptible individuals [23][24][25][23,24,25]. The availability of an animal model of MH, as certain breeds of pigs were found by chance to be susceptible to this anesthetic complication and to have an underlying muscle disease, provided additional evidence [18]. It has been demonstrated that overheating alone can trigger fatal MH in susceptible experimental piglets, thus supporting the association between MHS and heat stroke in humans and between MHS and sudden infant death syndrome, which may be due to overheating [18]. Some patients with environmental heat stroke have been found to have histories or family histories of MH or an association among MH-related genetic defects [26][27][28][29][26,27,28,29]. A rare case of a non-anesthetic, stress-induced hyperpyrexia death was described in a 12-year-old male who experienced an MH crisis during a humerus fracture operation and 8 months later presented MH followed by sudden death after exertion [27]. Furthermore, emotional stress may also cause or contribute to stress-induced MH [30][31][32][33][30,31,32,33].
4. Disorders Associated with Malignant Hyperthermia
The literature recognizes several myopathies associated with MHS and suggests several others (Table 1) [34][35][34,35].
Table 1.
Neuromuscular weakness classification for HM risk.
| Disease | Evidence in Adults | Evidence in Children | Suggested Perioperative Pathway |
|---|---|---|---|
| Upper motor neurons disease | |||
| Amyotrophic lateral sclerosis | None | None | standard |
| Myelin sheath disease | |||
| Multiple sclerosis | None | None | Standard |
| Guillain–Barré syndrome | None | None | Standard |
| Chronic inflammatory demyelinating polyneuropathy | None | None | Standard |
| Alexander disease | None | None | Standard |
| Krabbe disease | None | None | Standard |
| Adrenoleukodystrophy | None | None | Standard |
| Neuromyelitis optica spectrum disorders | None | None | Standard |
| Neuromuscular junction disease | |||
| Miastenia gravis | None | None | Standard |
| Muscular Dystrophy | |||
| Duchenne muscular dystrophy | Mild | Mild | Trigger-Free |
| Congenital muscular dystrophy | Mild | Mild | Trigger-Free |
| Facioscapulohumeral muscular dystrophy | Mild | Mild | Trigger-Free |
| Emery–Dreifuss muscular dystrophy | Mild | Mild | Trigger-Free |
| Becker muscular dystrophy | Mild | Mild | Trigger-Free |
| Channel disease | |||
| Myotonia congenita | None | None | Standard |
| Hypokalemic periodic paralysis | Strong | Strong | Trigger-Free |
| Central core disease | Strong | Strong | Trigger-Free |
| Cellular Metabolism disease | |||
| Mitochondrial disease | None | None | Standard |
| Kearns–Sayre syndrome | None | None | Standard |
| Glycogen storage disease | None | None | Standard |
| Lipid storage disorder | None | None | Standard |
| Other | |||
| Neonatal Palsy | None | None | Standard |
| Traumatic Damage | None | None | Standard |
Motor neuron diseases are one of the most implicated in this context. For example, amyotrophic lateral sclerosis and spinal muscular atrophy involve the degeneration of motor neurons, thus causing weakness, muscle atrophy, and spasticity, and therefore do not confer an increased probability of MHS [35].
Myelin sheath disorders are another series of disorders that may cause weakness but do not directly affect the muscle fiber and therefore do not increase susceptibility to MH [36]. However, areas of demyelination may be more prone to toxicity by local anesthetics; thus, GA may be preferred [36].
Autoimmune disorders (myasthenia gravis and Lambert–Eaton syndrome) are characterized by the presence of pathogenic antibodies directed against the acetylcholine receptor at the neuromuscular junction, and although they may cause abnormal responses to depolarizing and non-depolarizing neuromuscular blocking agents, they do not increase MHS [36][37][36,37].
Similarly, dystrophic and non-dystrophic myotonic syndromes, a broad class of rare multisystemic myopathies clinically characterized by a combination of myotonia (impairment of muscle relaxation after voluntary contraction), muscle weakness, wasting, and myalgia due to genetic defects which involve the muscular isoforms of various ion channels, have traditionally, albeit erroneously, been considered at increased risk of developing MH [35]. Patients with these myopathies have a chance of developing MH that is equivalent to that of the general population, with one exception, represented by hypokalemic periodic paralysis (HypoPP), in most cases caused by mutations in the skeletal muscle voltage-gated Ca2+ channel encoded by CACNA1S (HypoPP type 1) [36]. However, the latest research shows that myotonic patients with MH crisis can have mutations at two distinct genetic loci, one for myotonia and one for MHS [14][38][14,38]. Therefore, although episodes occurred without evidence of the MH hallmark of hypermetabolism, non-triggering anesthetics should be recommended to reduce the risk of rhabdomyolysis [38].
The dystrophinopathies, which include Duchenne and Becker muscular dystrophy, cover a spectrum of X-linked muscle diseases, usually presenting in early childhood, characterized by progressive proximal muscle weakness and muscle fiber degeneration [37][39][37,39]. In addition, there are some concerns about the risk of MH because, in these patients, phenomena of volatile anesthetic-induced rhabdomyolysis and hyperkalemia are described [37][39][37,39].
A very challenging category of neuromuscular syndromes is that of mitochondrial diseases [40]. Kearns–Sayre syndrome, mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes (MELAS), or Leigh syndrome could be manifested in many overlapping patterns, but pediatric onset is generally more severe [40]. Because patients with mitochondrial diseases demonstrate hypersensitivity to volatile anesthetics, many practitioners avoid using volatile agents [41]. However, evidence denies any connection between mitochondrial disease and MH [42]. On the contrary halogenated agents characterized by rapid elimination (sevoflurane, desflurane) can be used, but caution should be paid to the increased risk of developing propofol-related infusion syndrome [42].
Some reports put rare or very rare diseases and metabolic syndromes under the MH spotlight, although the literature does not support suspicions in most cases. For example, glycogen storage diseases are not considered at risk of MH due to unclear metabolic adverse events described in some reports [43]. All anesthetic agents have been used for GA in children with Pompe disease, but no complication could be clearly associated with clinical MH [44]. Similarly, McArdle’s disease has been considered in the MH group for two patients testing positive for the in vitro contracture test and atypical reactions [44]. McArdle’s patients are probably more susceptible to muscle cramps, which may cause diagnostic confusion [44]. Patients with Noonan syndrome share a similar phenotypic appearance with King–Denborough syndrome, including pterygium colli, down-slanting palpebral fissures, eyelid ptosis, short stature, and pectus excavatum, and have long been associated with an increased risk of MH [45][46][45,46]. However, the absence of proof emerging from the literature, along with knowledge of the genetic basis of the 2 disorders (mutation on chromosome 19 near the gene that encodes the ryanodine receptor in the King–Denbourough syndrome and on chromosome 12 for the Noonan syndrome) led to the exclusion of MHS in Noonan syndrome patients [45][46][45,46].
Myopathies clearly recognized to be associated with MH are masseter muscle rigidity (MMR), central core disease (CCD), multi-mini core disease (MmD), centronuclear myopathy, HypoPP, Native American myopathy (NAM), and King–Denbourough syndrome [6][7][8][9][10][12][47][6,7,8,9,10,12,47].
Succinylcholine-induced MMR occurs in 1 in 100 children after induction with inhaled anesthesia and succinylcholine administration, and the clinical incidence of MH after MMR is estimated to be 15% [48][49][50][48,49,50]. However, muscle biopsy reveals that 50% of patients experiencing MMR show MHS [49]. CCD refers to a rare non-progressive myopathy caused by an RYR1 mutation with mainly autosomal dominant inheritance, presenting in infancy, characterized by hypotonia and muscle weakness, sustained by a predominance of type I muscular fibers containing clearly defined areas (cores) lacking oxidative enzyme activity [51][52][53][54][51,52,53,54]. The mutation of RYR1 in CCD implies insufficient Ca2+ concentration in the cytoplasm, causes excitation–contraction decoupling, and finally leads to clinical muscle weakness [53][54][55][53,54,55]. These patients often demonstrate MHS, but MH and CCD phenotypes do not always co-segregate within families [56][57][56,57]. Patients with MH may present with cores despite being clinically asymptomatic and with some RYR1 variants specific to CCD. Although RYR1 variants are the most commonly identified cause of CCD, they show genetic heterogeneity [56][57][56,57]. MmD is an autosomal recessive, early onset congenital myopathy that strikes bulbar, respiratory, and extraocular muscles [58]. Some variants of RYR1 resulting in altered Ca2+ release from intracellular stores have been associated with MmD [59]. Currently, the most accredited hypothesis is that one subset of RYR1 variants may result in both MH and MmD while another may be associated only with MmD [6][11][6,11].