Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Houria Daimi and Version 2 by Rita Xu.
The endoplasmic reticulum (ER) is a principal subcellular organelle responsible for protein quality control in the secretory pathway, preventing protein misfolding and aggregation. Failure of protein quality control in the ER triggers several molecular mechanisms such as ER-associated degradation (ERAD), the unfolded protein response (UPR) or reticulophagy, which are activated upon ER stress (ERS) to re-establish protein homeostasis by transcriptionally and translationally regulated complex signalling pathways.
- non-coding RNAs
- ER stress
- UPR
- ERAD
1. Introduction
The endoplasmic reticulum (ER) is the largest, multifunctional, membrane-like, cellular organelle, composed of smooth and rough ER and forms an interconnected network of space [1]. ER exerts a pivotal role in three physiological cellular processes: (1) modulation of correct protein secretion, folding and translocation from ER lumen, (2) regulation of intracellular Ca2+ uptake, storage and signalling and (3) production of several membrane cellular lipids such as cholesterol, ceramides and/or glycerophospholipids [2][3][2,3].
A significant percentage of intracellular proteins are synthesised in ER lumen, wherein its oxidative environment facilitates the formation of disulphide bonds on proteins by different chaperones, foldases and cofactors. Generating disulphide bonds leads to proper secretory and transmembrane protein folding [4][5][4,5]. Alteration of ER protein folding capacity may cause an increased proportion of unfolded and misfolded proteins in ER lumen which triggers loss of ER homeostasis and proteostasis and generates a detrimental cellular environment [6][7][8][6,7,8]. Several molecular and biophysical mechanisms are triggered to reverse and restore ER homeostasis such as (1) ER-associated degradation (ERAD), which triggers the misfolded protein degradation from ER lumen; (2) Unfolded protein response (UPR) involving the restoration of ER proteostasis by activation of three transduction signalling –IRE1, ATF6 and PERK branch-; and (3) Reticulophagy, the process of ER remodelling by autophagy of membranes and associated proteins (see reviews [9][10][11][12][13][14][9,10,11,12,13,14]). Pathophysiological factors occurring in cardiovascular diseases (CVDs) such as metabolic derangement, hypoxia, hypertrophy or inflammation require an increased protein expression, thus enhancing the disruption of the cellular proteostasis [15][16][17][18][19][20][21][15,16,17,18,19,20,21]. As a consequence of the increased requirement of protein synthesis, ER homeostasis is ruptured and different subpopulations of cardiac cells suffer an unfolded and misfolded protein accumulation, which in turn, induces ER stress [22][23][24][25][22,23,24,25]. Accumulation of deleterious proteins triggers ER stress signalling which exerts a bivalent role both beneficial and/or harmful in cardiovascular function [26][27][28][29][30][31][32][33][26,27,28,29,30,31,32,33]. Furthermore, ER homeostasis is closely associated with normal cardiovascular function, and ER stress is considered a cause and a consequence of an extensive variety of CVDs such as ischaemic heart disease, hypertension, heart failure and dilated cardiomyopathy [34][35][36][37][34,35,36,37].
2. ERS and UPR Signalling
Since ER is crucial for the correct functioning of the cell, there are ER stress response mechanisms that control the degradation of the unfolded or misfolded proteins aiming to maintain ER homeostasis. The core mechanism of control is the activation of unfolded protein response (UPR). The central function of UPR is the inhibition of protein synthesis and the increase in the folding capacity of the ER. UPR may be activated by three different signal transduction pathways, initiated by three proteins located in ER membrane: inositol requiring protein 1 (IRE1), protein kinase RNA-like ER kinase (PERK) and activating transcription factor 6 (ATF6). In basal conditions, these molecules are bound to a chaperone named Bip (or GRP78) and remain attached to the ER membrane. However, when misfolded or unfolded proteins are accumulated, they dissociate and trigger three different signalling pathways induced by IRE1, PERK and ATF6 to resolve ER stress (Figure 1) [38][39][38,39].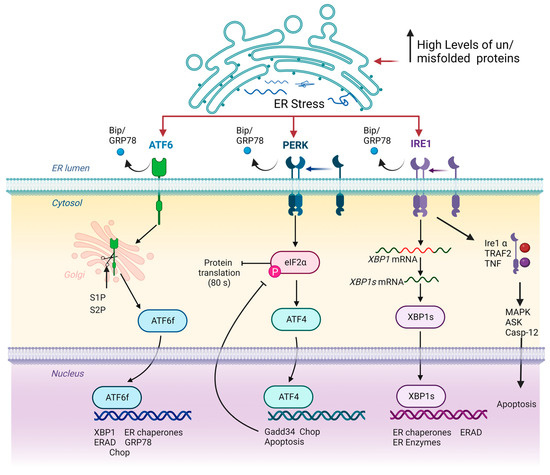
Figure 1. Schematic representation of UPR signalling pathways. Note that in homeostatic conditions, IRE1, ATF6 and PERK remain attached to ER membrane, exerting a sensorial cellular function. Loss of ER homeostasis by increased concentration of misfolded or unfolded proteins triggers the delivery of IRE1, ATF6 and PERK proteins to ER membrane by Bip/GRP78 factor. Subsequently, ATF6 is modified, acquiring transcriptional activity, while IRE1 and PERK activate ATF4 and XBP1s, respectively, which in turn exerts a transcriptional function. Inside the nucleus, ATF6, ATF4 and XBP1s initiate the expression of several genes that aim to restore cellular proteostasis. Arrows and bar-headed lines represent activation and inhibition effects respectively.
3. Role of ERS and UPR in Cardiovascular Diseases
ERS and subsequent activation of UPR exhibit both beneficial and deleterious effects in cardiovascular diseases, being thus considered both as a cause and consequence of them. Cardiac pathologies increase the demand and requirements of the ER function since an enhanced proportion of misfolded proteins triggers in many cases the loss of homeostasis of this organelle. Furthermore, ERS exerts a pivotal role in the modulation of both Ca2+ homeostasis and mitochondrial function in cardiomyocytes. Prola et al. (2019) have demonstrated that Tunicamycin (TM) treated cardiomyocytes display several changes in their cytoplasm ultrastructure, such as enlarged cytosol, decreased mitochondrial number, increased proportion of mitochondria-associated-membrane (MAM) fraction and expansion and dislocation of the ER near to nucleus and thus away from the sarcomeres. Accordingly, ERS reduced the mitochondrial number and function by downregulating several proteins involved in mitochondrial biogenesis such as PGC1a, TFAM, NRF1 or CS and thus is involved in the reduction of the mitochondrial capability to produce ATP [72]. Initially, adaptive UPR activation is capable of restoring ER and mitochondrial function and thus sustaining cardiac homeostasis. Curiously, the effects of molecular signalling pathways triggered by ERS are different within distinct cardiovascular injuries such as atherosclerosis, myocardial infarction, heart failure, cardiac hypertrophy or ischaemia and reperfusion (I/R) injury among others. For example, in heart failure or hypertrophy cardiac response caused by cardiac pressure overload, the PERK signalling pathway increases autophagy while it reduces ROS levels and apoptosis ratio by upregulation of EIF2A and ATF4, which in turn restores protein-folding capacity [31][73][31,73]. A sustained upregulation of the axis EIF2A-ATF4 will produce an increase in the cardiomyocyte apoptosis triggered by CHOP and these processes can influence the progression of cardiac diseases. In addition, PERK restores Ca2+ intracellular concentration by modulating Serca2a and Calreticulin, demonstrating its requirement for a proper ER-dependent ion homeostasis [74]. Unlike PERK, ATF6 is involved in the progression of cardiac hypertrophy and heart failure response thus exerting a harmful role. However, a protective role of ATF6 has been described in I/R injury suggesting a dependent and complex function of UPR based on the type of cardiac injury. Effects of UPRs’ downstream pathways have been elucidated using several murine models, which have highlighted the importance of ER stress and dependent molecular mechanisms in cardiac homeostasis and pathology (Figure 2). Curiously, ATF6 deficient mice display a worse cardiac function and recovery from infarction after (I/R) injury and increased damage with respect to controls [26]. Furthermore, ATF6 gain-of-function mice exhibits an alleviated myocardial infarction after I/R injury demonstrating that ATF6 is required to protect the heart from damage and injury caused by myocardial infarction [75]. Like ATF6, Xbp1s deficient mice display a worse recovery from heart failure showing an increased infarct size while in vivo overexpression of this gene is translated into reduced infarct size after I/R injury. Similar to that observed in ATF6 and Xbp1s overexpression mouse models, in vivo gain-of-function of Ire1 results in preserved cardiac function and reduced fibrosis after myocardial infarction [27][76][27,76]. Unlike IRE1 or ATF6, PERK deficiency has a beneficial phenotype after heart failure displaying protection against pressure overload myocardial infarction suggesting that while ATF6 and IRE1 exert a protective role against heart failure, PERK and its downstream pathways are detrimental [74]. Furthermore, PERK is a key gene involved in the transcription activation of CHOP, an essential factor to trigger ERS-associated apoptosis. CHOP-deficient mice are resistant to cardiac hypertrophy, increased fibrosis and cardiac dysfunction pinpointing the importance of apoptosis in deleterious processes related to cardiovascular diseases [77]. Furthermore, loss of function of enzymes related to ERAD signalling have been carried out, reflecting the importance of this mechanism in cardiovascular diseases. For example, Hrp1 deficient mice display an exacerbated cardiac dysfunction after myocardial infarction demonstrating that loss of one mechanism either UPR signalling or ERAD components is enough to impede recovery from cardiac injury [78].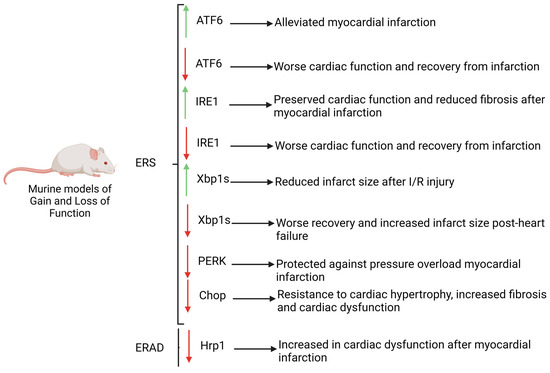
Figure 2. Murine models of gain and loss of function of key genes involved in UPR and ERAD pathways. Note that the deficiency of ATF6, IRE1, CHOP and HRP1 promotes the progression of CVDs, whereas low levels of PERK exert a protective role. Red arrows: downregulation, green arrows: upregulation.