Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Adrian Casas Benito and Version 2 by Jason Zhu.
Otto Warburg discovered that cancer cells use a fermentative rather than oxidative metabolism even though the former is more inefficient in terms of energy production per molecule of glucose. Cancer cells increase the use of this fermentative metabolism even in the presence of oxygen, and this process is called aerobic glycolysis or the Warburg effect.
- cancer
- metabolism
- Warburg effect
1. Cancer Cells Have a Different Metabolism
The main metabolic pathway used by most cells to obtain energy is oxidative phosphorylation, which includes the Krebs cycle, as it is the most efficient way to produce energy in the form of adenosine triphosphate (ATP). Glucose is the main biochemical fuel, which enters the cells through glucose transporters (GLUT) and undergoes glycolysis, providing pyruvate as a final product. Under normoxic conditions, although a small amount of pyruvate is transformed into lactate, most of it enters the mitochondrion where it is converted into acetyl-CoA, which is the main substrate for the citric acid cycle (CAC), also known as tricarboxylic acid cycle (TCA) or Krebs cycle. The main function of the TCA is the production of reducing power in the form of nicotinamide adenine dinucleotide (NADH) or flavin adenine dinucleotide (FADH2). These reduced molecules are the main electron donors for the mitochondrial electron transport chain (ETC) which generates more than 30 ATP molecules per molecule of glucose, making it the most important and efficient source of energy in the cell [1][11].
Almost one hundred years ago, in the decade of 1920, Otto Warburg for the first time proposed that cancer cells undergo a metabolic reprogramming that consists of two major features: (i) an increased glucose uptake and (ii) a metabolic switch to fermentative degradation of glucose incrementing lactate production (Figure 1). It is important to note that cancer cells present this special metabolic phenotype even when oxygen is available and that this type of metabolism is more inefficient when compared with the oxidative metabolism used by normal cells (Figure 1). Therefore, this phenomenon was dubbed aerobic glycolysis or the Warburg effect [2][3][12,13].
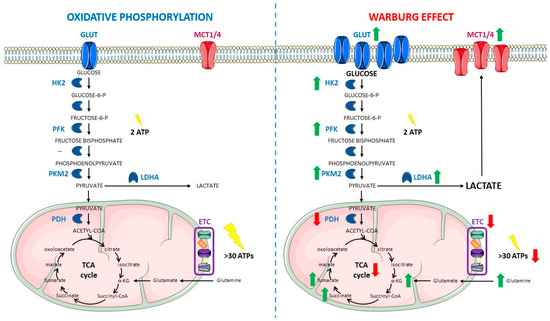
Figure 1. Comparison between oxidative phosphorylation (left) and the Warburg effect (right). Within oxidative metabolism, glucose is introduced inside the cell by Glucose Transporters (GLUTs) and degraded through glycolysis to provide pyruvate, which is mainly introduced inside the mitochondrion, with only a small amount being converted into lactate in the cytoplasm. Inside the mitochondrion, acetyl-CoA is obtained from pyruvate and enters the Tricarboxylic Acid (TCA) cycle to generate intermediates for the Electron transport Chain (ETC). Glutamine can be an important source of TCA α-ketoglutarate (α-KG) by entering inside the mitochondrion, being converted into glutamate and finally into the TCA intermediate. The whole process allows the cell to obtain more than 30 Adenosine Triphosphate (ATP) molecules per each glucose molecule catabolized. Under Warburg metabolism, GLUTs are overexpressed, which results in an increased glucose uptake. At the same time, there is an upregulation in the glycolytic enzymes Hexokinase 2 (HK2), Phosphofructokinase (PFK), and Pyruvate Kinase M2 isoform (PKM2), which leads to an increased glycolytic rate. The glycolytic rate may exceed the mitochondrial rate of pyruvate oxidation, thus making lactate accumulation unavoidable when glucose is so abundant inside the cells. Furthermore, Lactate Dehydrogenase A (LDHA) is also overexpressed in cancer metabolism, contributing to the generation of high levels of lactate. This accumulated lactate is pumped out of the cells through Monocarboxylate Transporters (MCT1/4) causing microenvironment acidification, which in turn triggers protumoral signaling pathways. In these conditions, Pyruvate Dehydrogenase (PDH) and the TCA cycle are also downregulated. At the same time, in several types of cancer, the coincidence of several mutations in the TCA cycle enzymes and the decreased entry of pyruvate into the TCA cycle further impair oxidative metabolism. The interruption of the TCA cycle decreases levels of malate and generates accumulation of some oncometabolites, such as fumarate, succinate, or α-KG. Under aerobic glycolysis, glutamine metabolism is enhanced, therefore contributing to α-KG accumulation as well. Now, the fermentative route provides less energy to the cell, obtaining the sum of 2 molecules of ATP per molecule of glucose in the glycolytic steps and a reduced amount of ATP molecules on this alternative and reduced oxidative phosphorylation. Green arrows indicate a net increase whereas red arrows show significant decreases. The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
Despite being a major finding, this information remained mostly unnoticed until a few decades ago, when molecular biologists’ studies associated some oncogenes with cancer cell metabolism, and the Warburg effect was put back in the spotlight for cancer scientists. According to Warburg’s initial hypothesis, cancer cells engaged in this differential metabolism because they were damaged and their mitochondrial respiration machinery was perturbed [4][14]. Warburg went as far as to argue that the cause of cancer is this “irreversible injuring in cell respiration” [4][14]. However, it is now clear that mitochondrial function is essential for cancer cell viability, because elimination of cancer cell mitochondrial DNA (mtDNA) reduces their growth rate and tumorigenicity [5][15]. Different studies over the past decades have demonstrated that human tumors also respire; in fact, oxidative metabolism persists in several types of cancer and may even exceed the respiration rate of adjacent non-malignant tissue [6][7][8][16,17,18], everything depends on the type of tumor. For example, Bartman et al. [9][19] found that TCA cycle flux is suppressed in five primary solid tumor models; including spontaneous pancreatic adenocarcinoma, syngeneic pancreatic adenocarcinoma allograft tumors, spontaneous lung adenocarcinoma, xenograft colorectal cancer tumors, and flank Non Small-Cell Lung Cancer allograft tumors; but it is increased in lung metastases of breast cancer relative to primary orthotopic tumors. Furthermore, some recent data suggest that the observed mitochondrial rewiring that occurs due to the Warburg effect in some cancer cells, but not in all of them, is the result of an adaptive function [10][11][20,21]. In this regard, some studies suggest lactic fermentation might not be compensating mitochondrial deficiencies, but it could be an alternative mechanism to provide fuel for ATP production and to maintain large pools of glycolytic metabolites to support anabolic metabolism [11][12][21,22]. Still, now, the reason why a more inefficient metabolism is selected by some cancer cells remains intriguing.
It is well known that normal cells grow and regulate their metabolism according to different growth signals and receptors. In cancer, cells proliferate in a growth signal-independent manner with an accelerated biomass production that greatly increases the cellular anabolic demand while providing extra by-products that might contribute to tumorigenesis. Apparently, these accumulated biochemical intermediates are needed to maintain the altered proliferative rhythm, thus generating a dependency on this less-efficient metabolism [13][14][15][23,24,25]. Cancer cell’s dependency on the Warburg effect might also end up inducing autophagy or apoptosis, as the nutrient supply needs to be quite high to maintain the increased needs of the cell through aerobic glycolysis. Overall, it may seem that choosing the Warburg effect over other more efficient metabolisms is not a smart choice for cancer cells. However, other factors need to be taken into account. Although ATP production efficiency is radically reduced when compared to oxidative phosphorylation, this is not usually a problem for cancer cells, as resources tend to be unlimited, at least in the early phases of the disease [16][17][26,27]. Moreover, cancer cell requirements, other than ATP production, might be better satisfied with this alternative metabolism. For instance, glycolytic ATP generation takes place at a much higher velocity than through oxidative phosphorylation, so the ratio ATP generation/time is increased with this metabolic reprogramming [18][19][28,29]. Another reason could be that glucose and glycolytic derivatives serve as precursors for fatty acids, non-essential amino acids, and nucleotide synthesis [20][30]. In addition, the products released by glycolytic cells such as lactate can serve as substrates for mitochondrial oxidative phosphorylation in neighboring cancer or immune cells [21][31].
However, aerobic glycolysis is not the only change observed in tumor metabolism. In addition to glucose, proliferating cancer cells also rely on glutamine as a major source of biomass synthesis and as a source of energy, with glutamine feeding into the TCA cycle [22][32], a process also known as glutamine addiction [23][33]. Glutamine is the most abundant non-essential amino acid in the bloodstream that can be supplied from blood or synthesized by glutamine synthetase (GS) to meet the increased metabolic needs of rapid proliferating cells at a concentration as high as 0.5 mM [24][25][34,35]. Glutamine is further metabolized with ammonia into glutamate by glutaminases (GLS1/2) [26][36]. Then, it typically refills the TCA cycle with α-ketoglutarate (α-KG) through the glutamine–glutamate–αKG pathway, catalyzed by GLS and glutamate dehydrogenase (GDH) (Figure 1), and finally producing GTP/ATP, NADPH, and pyruvate. This pathway is called glutaminolysis [27][37]. Reflecting the highly heterogeneous nature of cancer metabolism, the degree of glutamine dependence differs among cancers and there are certain cancer types more prone to glutamine addiction than others, such as glioblastoma (GBM), pancreatic, breast or lung cancer, among others [28][29][30][38,39,40]. Despite being a non-essential amino acid, glutamine is necessary for the survival of the cancer cells that are addicted to it [22][32] and that is probably the reason why cancer can cause significant changes in the way glutamine circulates in the body, significantly enhancing the release of glutamine into circulation by skeletal muscle via upregulation of GS [31][41]. Furthermore, there are data suggesting that increased glutamine metabolism in cancer cells also promotes tumorigenesis and cancer cell survival by maintaining redox homeostasis and ATP supply [22][23][32,33]. Regarding the glutamine addiction process in GBM cells, some studies suggest that lactate is formed as a carbon residue through glutamine-derived malate [10][11][20,21]. The same research group described the fact that, on this same pathway, alanine is also produced as a way to get rid of excessive nitrogen groups. This process takes place through alanine aminotransferase (ALT) or GDH action [20][30][32][30,40,42]. Alanine formed this way can be secreted into the medium where it can accumulate and may serve as additional fuel for other cancer cells [33][34][43,44].
Considering all these data, it seems obvious that cancer cells obtain several benefits from altering their metabolism, including the Warburg metabolism, thus suggesting an adaptive function, although this is probably not the only reason behind the metabolic switch observed in cancer.
2. What Are the Causes of the Warburg Effect?
Although cancer cells obtain some benefits from the Warburg effect, the exact reasons why cancer cells switch toward it remain unclear, as well as the timing of this switch. Though understanding the full exact mechanisms behind the Warburg effect is still a great challenge, some of them have already been identified and studied.
It is true that the mutagenic status and/or altered pathways, both characteristic of cancer cells, are crucial for the regulation of the Warburg metabolism. However, the high cellular heterogeneity present in tumors has made it difficult to characterize the effects of specific alterations and their role in the origin of the Warburg metabolism switch. For example, strong evidence exists that some mutations in TCA cycle enzymes, such as succinate dehydrogenase (SDH) and fumarate hydratase (FH), are associated to and impaired oxidative phosphorylation that highly contributes to the metabolic reprogramming observed in cancer cells [35][36][37][38][39][45,46,47,48,49], as well as with an increased hypoxia-inducible factor (HIF-1α)-mediated glucose uptake and glycolysis [40][41][42][50,51,52]. These mutations that take place in mitochondrial genes have been addressed as responsible for the Warburg effect’s features [43][53]. These mutations that have been described in various cancer types are responsible for altering mitochondrial metabolism, which in turn enhances tumorigenesis and permits cancer cell adaptation to challenging environments, such as the hypoxic ones [25][44][45][46][47][48][35,54,55,56,57,58]. This alteration in mitochondrial functions is what led Warburg to state that a mitochondrial dysfunction was observed in cancer cells [4][14]. However, it is now globally accepted that having functional mitochondria is essential for cancer cells [47][57], and the Warburg effect is now considered as an altered or hardwired mitochondrial metabolism due to metabolic reprogramming and not a malfunction of the mitochondria.
However, even tumors that have hijacked their metabolism to suppress pyruvate oxidation and produce lactate use residual or reprogrammed aspects of respiratory mitochondrial machinery and can rewire their mitochondrial metabolism to maintain pools of TCA-cycle metabolites that can be further used as intermediates for anabolism and to generate oncometabolites [11][49][50][51][21,59,60,61].
For some time, it was thought that overexpression of HIF-1α due to transient hypoxic conditions partially induced the Warburg effect. This theory was based on evidence showing a HIF-mediated increased glycolysis in tumors, where glucose transporters and glycolytic pathway enzymes where upregulated [52][62]. HIF-1α is the main sensor and regulator of the hypoxia response in the cell, as it is degraded in the presence of oxygen, whereas it accumulates and triggers hypoxia-induced signaling under low oxygen concentrations. HIF-mediated hypoxia response is similar to the Warburg effect´s one, as metabolic conditions and cell requirements are similar. That is why HIF is very important in cancer fermentative metabolism as a key regulator of the process, even in the presence of oxygen, and that is why this process is called pseudohypoxia. Interestingly, lactate dehydrogenase A (LDHA) and pyruvate dehydrogenase kinase 1 (PDK1) expressions are increased by HIF upregulation, thus inducing an accumulation of lactate and reducing the supply of pyruvate to the mitochondrion [53][63]. At the same time that HIF provokes a decrease in mitochondrial respiration, it also optimizes the efficiency of glycolysis by regulating cytochrome C oxidase isoform ratios [54][64]. Nevertheless, some studies identified the Warburg effect´s onset prior to hypoxia on several tumors [55][56][65,66]. Therefore, it seems that HIF regulation of the Warburg effect takes place after the latter has already been activated by oncogenic pathways, such as phosphatidylinositol-3 kinase (PI3K) or Mitogen-Activated Protein Kinase (MAPK), and also by stabilization due to oncometabolite-induced prolyl hydroxilase domain (PHD) protein inactivation [57][67].
As stated in the beginning of this section, just as with all other cancer hallmarks, the Warburg effect’s cause lays, ultimately, in gene mutations. For instance, the highly cancer-related pathway PI3K-Akt-mTOR induces an enhanced glucose capture, modulating GLUT1 expression and activating phosphofructokinase (PFK) and hexokinase (HK), among other enzymes, which contribute to high availability of glucose for the tumor cells, thus feeding the high needs of the Warburg metabolism [20][30]. Furthermore, HIF-mediated miR-199a5p inhibition leads to HK2 modulation and further increases on glucose availability [58][68]. Moreover, cancer cells express dimeric pyruvate kinase M2 isoform (PKM2) and upregulate PDK1, both of which inhibit pyruvate dehydrogenase (PDH), block pyruvate entrance into the mitochondrion to feed the TCA cycle, and result in an increased lactate production [59][60][69,70]. An increased AMP-activated protein kinase (AMPK) or c-MYC signaling, very common in cancer cells, also enhance the glycolytic flux through GLUT isoforms regulation or by inducing PFK or HK overexpression. The overexpression of glycolytic-involved genes (including LDHA, HK, GLUT1 or PKM2) has been shown in 24 types of cancer, including more than 70% of the total amount of human cancer cases [61][62][63][71,72,73]. At the epigenetic level, histone deacetylase (HDAC) inhibition has been shown to induce GLUT overexpression [64][74], and some sirtuins have been associated to Warburg’s metabolic switch through chromatin state regulation [65][75].
As previously mentioned, mutations in mitochondrial enzymes also play an important role in Warburg’s phenotype acquisition. Some examples include mutations in SDH, isocitrate dehydrogenase (IDH), and FH. What characterizes the Warburg effect is the lack of proportion between glycolysis and cellular respiration. Tumor cells that have switched towards a Warburg phenotype uptake high amounts of glucose in a rapid way and convert them into lactate even in the presence of oxygen. The proposed reasons to explain the aerobic glycolysis phenomenon are (1) that the glycolytic rate exceeds by far the maximal rate of mitochondrial pyruvate oxidation so lactate accumulation and secretion is unavoidable when glucose is so abundant [66][76]; (2) the overexpression of LDHA [67][68][77,78]; and (3) that due to mutations, the expression of some of the main TCA cycle enzymes is significantly decreased, thus reducing the entry of pyruvate into the TCA cycle and causing an impaired oxidative phosphorylation [35][45]. Ultimately, these mutations and the loss of oxidative metabolism contribute to oncometabolite accumulation (mainly succinate, fumarate, and α-KG), which is another important characteristic of aerobic glycolysis [57][67].
All these factors, and some others, such as the influence of tumor microenvironment (TME) components, the resulting acidosis, or mtDNA mutations, contribute to the metabolic switch [69][70][71][79,80,81]. However, this switch is not fully understood yet; there are gaps in the current knowledge of what initial changes lead to the Warburg status in cancer cells, mainly due to the high mutagenic variability observed in the cancer cells. Considering all this, the Warburg metabolism initiation needs further investigation that may lead to the development of new preventive or therapeutic strategies against cancer.
3. Main Features of the Warburg Effect
Although the reasons behind the switch from an oxidative metabolism to an aerobic glycolysis remain partially obscure, the Warburg effect has been extensively studied by now and its main characteristics and features have already been described.
As mentioned above, one of the main Warburg effect hallmarks is glucose uptake enhancement, mainly mediated by the increase in GLUT expression, meaning cancer cell survival also depends on the medium contents in nutrients capable to enter TCA cycle (that is why having alternative sources of carbon to enter the TCA cycle such as glutaminolysis-derived α-KG is so important for cancer cells). This upregulated acquisition is accompanied by an increase in glycolytic enzymes and a consequent increased glycolytic rate [67][77]. Moreover, pyruvate is not mainly metabolized to acetyl-CoA, as usual, but is diverted into lactate production, which is one of the main products responsible for driving the Warburg-associated increase in malignancy potential. As stated above, the upregulation of LDHA and PDK1, or mutations in these genes, are very important to induce the fermentative pathway [67][77]. Furthermore, glutaminolysis also contributes to lactate accumulation, which is responsible for acidification of the TME, which is important for tumor malignancy. This acidification of the medium happens because the cell tries to alleviate lactate accumulation in the cytoplasm through upregulation of the monocarboxylate transporter (MCT-4) (Figure 1), which ejects lactate from the cell, a process that is also regulated by HIF-1α [72][82]. However, these transporters work as symporters and, at the same time, they eliminate lactate and eject protons into the extracellular space, generating its acidification [73][74][83,84]. Lactate externalization has two more consequences: the first one is that extracellular lactate is internalized by other cancer or tumor microenvironment (TME) cells that express MCT-1. Then, those cells use the excess of lactate to trigger mitochondrial oxidative metabolism, generating a process known as the reverse Warburg effect, becoming more efficient in energy production [75][85]. The second consequence is that lactate can bind to G protein-coupled receptor 81 (GPR81) which, in turn, activates certain pathways such as PI3K/Akt, favoring cancer progression [76][86].
Another Warburg effect hallmark, and a general cancer trait, is the enhancement of anaplerotic reactions. The accelerated metabolism and the increased use of some molecular component sources make it necessary to induce additional anabolic pathways in order to regenerate Krebs cycle intermediates. Some of the enzymes relevant for these pathways are pyruvate carboxylase, aspartate aminotransferase (which restores oxaloacetate levels which are important for citrate synthesis), propionyl-CoA carboxylase (which functions in odd-chain fatty acid utilization in order to generate succinyl-CoA), or glutaminase (which intervenes in glutamine catabolism, generating α-KG) [77][87].
All these changes in metabolism not only allow cancer cells to obtain more energy, but are also linked with the upregulation of protumoral signaling pathways, helping cancer cells to gain malignancy. This is another reason why the Warburg effect might constitute a selective advantage for cancer cells; this metabolism enhances the availability and function of certain metabolites that promote cancer progression, and that is why they are called oncometabolites. These molecules usually have a specific role in metabolism, but their excessive accumulation under Warburg conditions help cancer progress. The best known oncometabolite is lactate, whose pivotal role in cancer progression has been already summed up in an extensive review [72][82]. Other important oncometabolites include 2-hydroxyglutarate, fumarate, and succinate (Figure 1) [78][88]. However, the role of other TCA-cycle metabolites in cancer initiation and progression should not be disregarded, even if they are not considered oncometabolites, and investigating them may open new and interesting topics in the research against cancer. A detailed review about this can be found in the work of Eniafe and Jiang [79][89].
The hexosamine biosynthetic pathway (HBP) generates UDP-GlcNAc, another oncometabolite. This pathway is also upregulated in cancer under the Warburg effect and both UDP-GlcNAc, its final product, and some intermediates are able to modulate signaling pathways that favor tumor progression. The HBP works as a metabolic sensor, so when more nutrients are present, both glycolytic and HBP pathways are enhanced [80][90].
Another relevant feature that occurs in the Warburg metabolism concerns reactive oxygen species (ROS). The role of ROS in cancer has been studied for many years, and it is widely known that cancer cells have a higher production of ROS. In addition, when metabolism is enhanced, especially in those cells that still maintain oxidative phosphorylation, ROS production rises further. To deal with this oxidative stress, the pentose phosphate pathway (PPP) uses glycolytic intermediates, which are hijacked from glycolysis, to generate NADPH as a protection against the increased amount of tumor-generated ROS [81][91].
Some of the previously described features, as well as other characteristics, have been useful to understand not only cancer metabolism, but also cancer behavior in general, which can ultimately impact clinical cancer management. For instance, based on the increased glucose consumption rates by cancer cells, Fluorodeoxyglucose (FdG) is commonly used for positron emission tomography (PET) imaging, which allows to diagnose and follow up tumors [52][82][62,92].
4. Classical Approaches Targeting the Warburg Effect to Fight Cancer
Characterizing cancer metabolism is not only important to cancer diagnosis, but it may also help in cancer therapy development by targeting specific metabolic cancer cell features. This is why having a better understanding of Warburg metabolism and its main characteristics, such as accumulated oncometabolites, is so important, as it may represent a new avenue of research to discover new anti-cancer strategies.
In an attempt to impair cancer metabolism by targeting the Warburg effect characteristics, some treatments have already been tested. Some approaches in this line include the inhibition of glycolytic enzymes; for instance, HK2 inhibitors, such as lonidamine, have reached clinical trials in combination with other antitumoral compounds, although no significant effects were obtained or toxicity appeared [83][93]. 3-bromopyruvate is another HK2 inhibitor that has shown good preclinical results in liver and prostate cancer [8][84][18,94]. The use of GLUT inhibitors is also a common approach. For instance, WZB117 increased the effect of other treatments, while STF-31 showed positive preclinical results [85][86][87][95,96,97]. Another way to inhibit the Warburg effect is targeting lactate production by LDHA-mediated approaches. Some examples are urotilin M6 and FX11, which have been proven to inhibit LDHA in preclinical studies on hepatocytes and adipocytes, and on 15 patient-derived TP53-mutated mouse xenografts, respectively [8][87][88][89][90][18,97,98,99,100]. MCT inhibition is also an interesting way to attack cancer metabolism, and AZD-3965 is a good example of an MCT1 inhibitor that is currently in clinical phase of research for advanced cancers [87][91][92][97,101,102]. Many HIF inhibitors have also attracted great interest in anti-cancer therapy research. Among these, two synthetic molecules, PT2385 and PT2399, have shown good results in renal carcinoma cell lines [8][93][18,103]. Other attempts include pyruvate kinase M2 inhibition [94][104] or ketogenic diets [95][105]. It is true that in the past decades have been some controversy about the beneficial role of ketogenic diet in human health [96][97][98][106,107,108], however, in the specific case of cancer this type of diet targets the Warburg effect in tumor cells [99][109]. The characteristics of ketogenic diet, a high-fat, low-carbohydrate diet with adequate amounts of protein, creates an unfavorable metabolic environment for cancer cells by drastically reducing the amount of glucose available for tumor cells [99][109]. Furthermore, in response to an increase of ketone bodies under ketogenic diet feeding, CD4+ and CD8+ T cells are metabolically reprogrammed to rely on OXPHOS and they are able to secrete more cytokines [100][110]. In addition, it appears to sensitize most cancers to standard treatment by exploiting the reprogramed metabolism of cancer cells and by boosting effector T cell functions, making this diet a promising candidate as an adjuvant cancer therapy [95][100][105,110].
Some possible molecules to target glutamine metabolism have also been developed. One candidate is CB-839, under the commercial name Telaglenastat, which has proven to sensitize cancer cells to radiation in cervical cancer and in head and neck squamous cell carcinoma, in in vivo models, and has even reached clinical trials against some tumors. Other approaches are L-asparaginase (L-ASP), which induces glutamine depletion and increases radiosensitivity and induces cell cycle arrest in ARCaPM cells, or JHU083 and DRP-104, which are general glutamine inhibitors [88][92][101][102][98,102,111,112].
Another good option is targeting the Warburg phenotype by tackling more upstream actors in the metabolic pathway. For example, sirtuin 6 (SIRT6) is a tumor suppressor that inhibits HIF-1α and, as a consequence, reduces HK, LDHA and GLUT1 [65][103][75,113].
Recently, the different complexes of the ETC have attracted interest as a possible target to develop new therapies to compensate or reverse the mitochondrial switch observed in cancer [16][104][105][26,114,115]. In this regard, the work by Moreira et al. [106][116] not only summarizes the fundamental role of cytochrome c oxidase, complex IV of the ETC, in the effectiveness or not of chemotherapy, immunotherapy and probably radiotherapy treatments; but it also provides new interesting anti-cancer metabolic therapy strategies to hijack Warburg phenotype. They proposed that photosensitizers such as methylene blue, chlorophyll, and protoporphyrin could play an intermediary role in the electron decongestion of ETC by catalyzing the activation of 3O2 into 1O2 and thus promoting apoptosis by accumulation of ROS species in tumoral cells, especially those that are resistant to conventional treatments [106][116].
Unfortunately, despite the great effort made in the last decade to target cancer metabolism, the clinical applications are still modest. Cancer cell plasticity is enormous, and that enables them to compensate the lack of a specific pathway by obtaining energy through other means. For example, glycolysis inhibition can be compensated by glutaminolysis [107][108][117,118], as glutamine drives the glucose-independent TCA cycle, as described above.
So far, it has been difficult to obtain enough specificity without reaching high levels of toxicity with the previously explained options [109][119]. A potential alternative for these problems is the use of combined therapies to avoid resistance, for instance, by simultaneously inhibiting glycolysis and glutaminolysis. Another way would be finding new methods to deliver the drug specifically on the tumor site to avoid toxicity in normal tissues and perhaps increase the local dosage.
Another problem for these therapeutic options is that not all tumor cells rely on aerobic glycolysis; for instance, cancer stem cells and other slow-proliferating cells are very resistant to these treatments because of their lower growth rates and different metabolism. That is probably the reason why very few Warburg-based treatments have reached clinical phases [8][18]. However, targeting the Warburg effect or its main features, including its associated oncometabolites, is still a promising alternative for anti-cancer therapy research. More investigation is needed to properly address the challenges of this still mostly unexplored route.