Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Natalia Stepanova and Version 2 by Dean Liu.
Oxalate is often referred to as an “anti-nutrient” because of its ability to bind with minerals and form various solubility complexes. Specifically, oxalate is a negatively charged ion that forms crystals by binding with positively charged ions, such as calcium, magnesium, iron, zinc, sodium, and potassium.
- oxalate
- chronic kidney disease
- hemodialysis
- peritoneal dialysis
1. Oxalate Implications in Health and Disease
Oxalate is often referred to as an “anti-nutrient” because of its ability to bind with minerals and form various solubility complexes. Specifically, oxalate is a negatively charged ion that forms crystals by binding with positively charged ions, such as calcium, magnesium, iron, zinc, sodium, and potassium [1][2][32,33]. The solubility of these salts varies widely, with sodium and potassium oxalate being soluble and calcium, magnesium, zinc, iron, and other cations forming as less soluble to practically insoluble oxalate compounds [2][33]. Specifically, the solubility product constant (Ksp) values at 25 °C for these compounds are as follows: calcium oxalate (CaC2O4) with a Ksp of 2.7 × 10−9, magnesium oxalate (MgC2O4) with a Ksp of 8.5 × 10−5, zinc oxalate (ZnC2O4) with a Ksp of 2.7 × 10−8, and iron(II) oxalate (FeC2O4) with a Ksp of 2 × 10−7 [3][34]. This binding capacity of oxalate with minerals can cause mineral deficiencies and is associated with the formation of calcium oxalate kidney stones, thus earning the term “anti-nutrient” [2][4][33,35].
Oxalate is synthesized by a variety of cells, including liver cells, kidney cells, epithelial cells, and apocrine cells [5][6][3,36]. However, despite being a byproduct of cellular metabolism, the physiological functions of oxalate in the human body remain largely unknown. Although it was traditionally believed that circulating oxalate has no function in the human body, early studies have described three potential physiological roles. First, oxalate plays a role in the proximal tubule of human kidneys, where it is transported by the membrane transporter SLC26A6. This transport process has been found to stimulate the absorption of chloride, water, and sodium [7][8][14,37]. The second role is related to the production of H2O2 by oxalate oxidase, which can enhance the burst of phagocytes [9][38]. Finally, uracil and orotic acid, which are essential components of RNA involved in protein synthesis and pyrimidine nucleotide synthesis, respectively, require oxalate for their formation in human metabolism [5][3].
In trace amounts, oxalate is generally considered physiologically inert and is excreted from the body without clinical significance [10][11][4,5]. However, a decrease in glomerular filtration rate (GFR), as well as an increase in hepatic oxalate production or gastrointestinal oxalate absorption, can raise POx concentrations and result in increased UOx excretion, thereby elevating the risk of various pathological conditions [10][11][12][4,5,39]. Calcium-oxalate (CaOx) urolithiasis, primary hyperoxaluria, and oxalate nephropathy caused by either high oxalate intake or enteric hyperoxaluria are among the most thoroughly researched conditions [2][12][13][33,39,40]. Besides, elevated plasma and/or urinary oxalate have been linked to a number of other pathologic conditions, such as diabetes mellitus and obesity [14][41], autism [15][42], atherosclerosis [16][43], cardiovascular events [17][44], and neurological disorders [12][39]. Patients with primary hyperoxaluria are especially prone to oxalosis, a condition where CaOx deposits have been found in various extrarenal tissues, primarily in the heart, smooth muscle cells of vessels, bones, skin, and other organs [18][45]. However, oxalate deposition has also been reported in non-primary hyperoxaluria patients affecting extrarenal tissues such as the breast [19][46], lungs [20][47], thyroid gland [21][48], prostate [22][49], synovial fluid [23][50], and vascular tissues [24][51]. Furthermore, studies have suggested that oxalate may promote the proliferation of cancer and metastatic cells, contributing to the development of breast and prostate cancer [6][25][26][36,52,53]. The diversity of pathological conditions associated with oxalate, notwithstanding the vital role played by the kidneys and intestine in maintaining oxalate homeostasis, underpins the significant oxalate implications in the development and progression of chronic kidney disease (CKD).
2. The Interplay between Oxalate and CKD: A Vicious Cycle of Shared Risk Factors
CKD is a significant public health problem worldwide, as affected individuals are at increased risk for end-stage kidney disease (ESKD), cardiovascular disease, and a wide range of CKD-related mental and physical illnesses, leading to premature mortality [27][28][54,55]. Oxalate has been shown to be associated with CKD progression [29][30][56,57], CKD- and ESKD-associated cardiovascular diseases [31][32][33][34][58,59,60,61], polycystic kidney disease progression [35][62], and/or poor renal allograft survival [35][36][62,63]. In CKD of any cause, impaired kidney function and associated gut dysbiosis can cause a buildup of plasma and glomerular ultrafiltrate oxalate, increasing oxalate exposure of the remaining tubule cells and decreasing the renal clearance of oxalate, leading to its accumulation in the body [10][11][37][4,5,28]. Oxalate retention can initiate a vicious cycle of progressive kidney damage and a further decline in GFR. In turn, the formation of oxalate crystals in the kidneys can accelerate inflammation and scarring, leading to a decline in kidney function and contributing to the progression of CKD [29][38][56,64]. This interplay forms a vicious cycle between oxalate and CKD with shared risk factors, where each condition exacerbates the other by promoting the accumulation of oxalate in the body, ultimately leading to an increased risk of CKD progression (Figure 12).
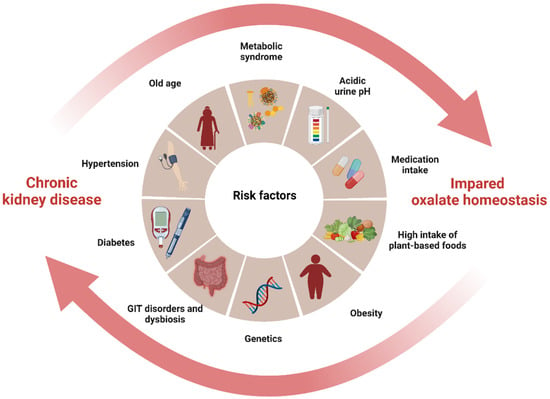
Figure 12. The interplay between impaired oxalate homeostasis and CKD (created with BioRender.com). The diagram illustrates the interplay between impaired oxalate homeostasis and CKD by highlighting the shared risk factors that contribute to both diseases and pointing to their common origin. The cycle formed by these pathological conditions mutually exacerbates each other, leading to an accumulation of oxalate in the body and subsequently increasing the risk of CKD progression.
Specifically, it has been observed that an imbalanced intake of oxalate and calcium in the diet is associated with a significant increase in the risk of developing hypertension [39][65], which is a strong risk factor for both impaired oxalate homeostasis and CKD through its damaging effect on the renal microvasculature. In obesity, there is an increased risk of developing both insulin resistance and hypertension, which can contribute to impaired oxalate homeostasis and CKD [14][40][41,66]. Similarly, metabolic syndrome, which includes insulin resistance, dyslipidemia, hypertension, and obesity, is independently associated with both CaOx nephrolithiasis and CKD [41][42][67,68]. Furthermore, a mutation in the SLC26A6 oxalate transporter has been linked to enteric hyperoxaluria and nephrolithiasis, indicating the possibility of an inherited form of this condition [43][69]. Although there is limited information on the relationship between SLC26A6 mutation and CKD, this anion transporter plays an important role in kidney salt absorption, acid-base balance, vascular volume homeostasis, and blood pressure regulation, all of which contribute to the advancement of CKD [44][70]. Acidic urine pH (<5.0) is also associated with significantly increased UOx and protein excretion, resulting in CKD progression [45][46][71,72]. While it is generally believed that urine pH has little impact on the solubility of CaOx, some studies have shown that CaOx monohydrate crystals are likelier to form at pH 4.0 and less likely to form at pH 8.0, indicating a potential influence of urinary pH [47][73]. Moreover, changes in urinary pH can affect the excretion of specific substances such as citrate, which has been found to inhibit the formation of CaOx. Consequently, alterations in urinary pH can disrupt the balance of citrate, potentially leading to hyperoxaluria and contributing to the progression of CKD. Notably, CKD often presents with hypocitraturia and/or metabolic acidosis, which are characterized by low urine pH [48][74]. As a compensatory mechanism for the loss of nephrons and impaired overall acid excretion, there is an increase in acid excretion per nephron, which further promotes damage to the tubulointerstitial region and contributes to the progression of kidney disease [48][74].
Finally, dysbiosis of the gut microbiota (described below), as well as the use of nephrotoxic medications or medications that affect the gut microbiome and metabolome, have been shown to contribute to both increased oxalate burden and CKD [49][50][51][21,26,75]. Proton pump inhibitors, for example, are known to cause intestinal permeability, leading to both urolithiasis and CKD progression [52][53][76,77]. Antibiotics, high-risk nephrotoxicity drugs, can alter the gut microbiota, reducing the abundance of either ODB or their ability to degrade oxalate, increasing the risk of oxalate burden [49][50][21,26]. Similarly, vitamin D deficiency, a nearly common feature in CKD, was also shown to impact gut microbiota diversity, causing intestinal barrier dysfunction [54][78]. Additionally, a deficiency in vitamin D may lead to elevated levels of parathyroid hormone, which can stimulate bone resorption and cause hypercalcemia and hypercalciuria, potentially increasing the risk of hyperoxaluria [55][79].
3. Oxalate’s Role in the Pathogenesis of CKD: From Silent Culprit to Active Player
While oxalate has long been known to be a risk factor for kidney stones, its involvement in the pathogenesis of CKD has only recently come into focus. For many years, oxalate was considered a “silent culprit” in CKD, with its effects on kidney function thought to be limited to the formation of kidney stones. The primary mechanism for CKD from kidney stones is usually attributed to obstructive uropathy or recurrent kidney stone disease experience [56][57][80,81]. In fact, oxalate may play a much more active role in the development and progression of CKD, contributing to kidney damage beyond the formation of kidney stones.
The mechanisms involved in kidney damage caused by oxalate are multifactorial and complex, and their complete understanding is still lacking. The primary way by which oxalate can contribute to CKD is through the impairment of mitochondrial function [58][82]. Mitochondria are energy-producing organelles within cells, and disruption of mitochondrial function has been implicated in a number of disease processes, including CKD [59][60][83,84]. Oxalate can trigger mitochondrial dysfunction in renal epithelial cells through various mechanisms, such as impairing the mitochondrial respiratory chain, increased reactive oxygen species (ROS) generation and inflammation, disrupting the mitochondrial membrane potential, and affecting mitochondrial biogenesis [58][61][62][82,85,86]. In endothelial cells, POx at uremic concentrations alters intracellular calcium levels, increases the production of ROS, and promotes cell apoptosis, leading to oxidative stress and inflammation [63][64][87,88]. Oxalate-induced oxidative stress and inflammation lead to the activation of various signaling pathways, such as nuclear factor kappa B (NF-κB), mitogen-activated protein kinases (MAPKs), and NLRP3 inflammasome, and promote the production of proinflammatory cytokines and chemokines, such as interleukins (IL) -1β, -6, tumor necrosis factor α (TNF-α), monocyte chemoattractant protein-1 (MCP-1), and transforming growth factor β1 (TGF-β1) in renal epithelial cells [33][63][65][66][1,60,87,89]. In turn, the activation of the TGF-β signaling pathway induces extracellular matrix (ECM) synthesis and deposition, which can lead to fibrosis and scarring in the kidneys [67][68][90,91]. Oxalate can increase MCP-1 and TGF-β expression and activate downstream signaling pathways, leading to the upregulation of ECM proteins, such as collagen and fibronectin [67][68][90,91]. In addition, oxalate can induce apoptosis of renal epithelial cells through the activation of various apoptotic signaling pathways, such as caspase-3 and -9, and the Bcl-2 family of proteins [69][92].
At the systemic level, oxalate has been shown to stimulate human monocytes and promote the production of proinflammatory cytokines such as TNFα, IL-1β, IL-6, and IL-8 in vitro [70][93], leading to systemic low-grade inflammation, which is a significant contributor to the progression of CKD [71][72][73][94,95,96]. Additionally, oxalate can compromise macrophage metabolism, disrupt redox homeostasis, and alter cytokine signaling, resulting in a reduced antibacterial response and increased risk of infection [66][89]. Altered oxalate metabolism has also been shown to promote atherosclerosis by dysregulation of redox status, enhance inflammatory response, and affect cholesterol metabolism both in vitro and in vivo using genetically modified mice lacking the alanine-glyoxylate aminotransferase enzyme and the apolipoprotein E gene (apoE−/−) [16][43]. In the CKD model induced by a high-oxalate diet in C57BL/6 mice, the majority of CKD-related complications have been observed, including mineral bone disease, dyselectrolytemia, metabolic acidosis, arterial hypertension, and even cardiac fibrosis [33][60]. In the uremic atherosclerosis model using apoE−/− mice, a significant increase in aortic oxalate levels and serum levels of oxidative stress and inflammatory markers have been observed, suggesting a significant role for hyperoxalemia in promoting oxidative stress and systemic inflammation [63][87].
Overall, it is becoming increasingly clear that oxalate may play a much more active role in the pathogenesis of CKD beyond its traditional association with kidney stone formation. Oxalate can directly impact renal epithelial cells, activate various signaling pathways involved in inflammation, fibrosis, and apoptosis, and contribute to systemic low-grade inflammation. While the current understanding of the mechanisms by which oxalate contributes to CKD has improved, further research is necessary to fully comprehend its impact on kidney health.
4. Gut-Kidney Axis in CKD Oxalate Homeostasis
The gut–kidney axis is essential in maintaining oxalate homeostasis in CKD [11][38][74][75][5,31,64,97]. Despite extensive research, the precise mechanisms by which the kidneys and gut communicate to regulate oxalate homeostasis in CKD remain incompletely understood [74][31]. Nonetheless, it is evident that various conditions associated with secondary hyperoxaluria, such as Crohn’s disease, chronic pancreatitis, Roux-en-Y gastric bypass, and antibiotic use, contribute to the progression of kidney disease [18][37][28,45]. These conditions are linked to alterations in the gut microbiota and enteric oxalate handling, disrupting oxalate homeostasis and leading to hyperoxaluria/hyperoxalemia [11][74][76][77][5,18,31,98]. For example, patients with inflammatory bowel disease have been shown to have elevated enteric oxalate levels and reduced oxalate-degrading gene expression caused by the loss of O. formigenes, resulting in hyperoxaluria [78][19].
CKD, on the other hand, is a widely discussed cause of gut microbiota dysbiosis characterized by an increase in proteolytic bacterial populations, which produce uremic toxins such as ammonia, phenols, and indoles, and a decrease in saccharolytic populations that form short-chain fatty acids (SCFAs) [75][79][97,99]. CKD-associated gut microbiota dysbiosis can impact oxalate homeostasis directly (via a decrease in bacterial ODA) or indirectly (via compromising intestinal barrier function). In theour previous report, researcherswe showed that ODA in fecal microbiota has a direct association with the percentage of renal interstitial fibrosis in rats following glycerol-induced acute kidney injury over 70 days [80][100]. In addition, total fecal ODA has been found to be significantly lower in patients with ESKD compared to healthy controls [81][82][101,102]. Interestingly, it appears that ODA, rather than the quantity of ODB in the gut microbiota, may have an impact on oxalate homeostasis [49][82][21,102]. This hypothesis was supported by findings on the inverse association between ODA in fecal microbiota and serum indoxyl sulfate, UOx, and POx concentrations [81][82][101,102]. However, further investigation is required to confirm these findings.
Intestinal dysbiosis and compromised intestinal barrier function can facilitate the translocation of bacterial products and uremic metabolites from the gut lumen to the bloodstream, contributing to oxidative stress, chronic inflammation, and CKD progression [79][83][84][99,103,104]. Uremic metabolites such as indoxyl sulfate, p-cresyl sulfate, and their precursors have been shown to directly promote CaOx crystal production in vitro [85][105], while SCFAs reduced UOx excretion possibly by the regulation of the expression of SLC26A3/6 transporters [86][106]. Notably, the SLC26A3 transporter, a crucial chloride-bicarbonate exchanger responsible for the reabsorption of oxalate from the intestinal lumen into the bloodstream, plays a significant role in maintaining the integrity of the intestinal barrier [87][107]. The deletion of the DRA protein, an isoform of the SLC26A3 transporter, in knockout mice has been found to lead to a notable decrease in UOx and POx concentrations [88][108].
The presence of chronic inflammation caused by dysbiosis further exacerbates the decline in GFR, leading to reduced renal clearance of oxalate and the development of hyperoxalemia. In response to hyperoxalemia, enteric oxalate excretion becomes an important compensatory mechanism for maintaining oxalate homeostasis [38][89][90][2,13,64]. It has been hypothesized that in CKD, the intestine may adapt and increase the secretion of oxalate into the intestinal lumen by upregulating the expression of oxalate transporters [38][91][15,64]. A recent study on animal models of CKD found that the SLC26A6 oxalate transporter, which plays a key role in restricting net absorption by back-secreting oxalate into the lumen, is upregulated not only in the small intestine but also in the colon, resulting in increased oxalate removal through fecal excretion [91][15]. Controversially, reduced fecal oxalate excretion and increased POx concentration were observed in SLC26A6 gene-deficient mice, suggesting that enteric oxalate secretion via SLC26A6 plays a vital role in defending against hyperoxalemia in CKD [91][15]. It is essential to note that this is the only study on intestinal SLC26A6 expression in CKD models, and much work remains to be done to improve theour understanding of the impact of the gut-kidney axis on oxalate homeostasis in CKD. Although a definitive understanding of this complex relationship remains incomplete, Figure 23 provides a schematic summary of current knowledge on the gut–kidney axis in CKD oxalate homeostasis.
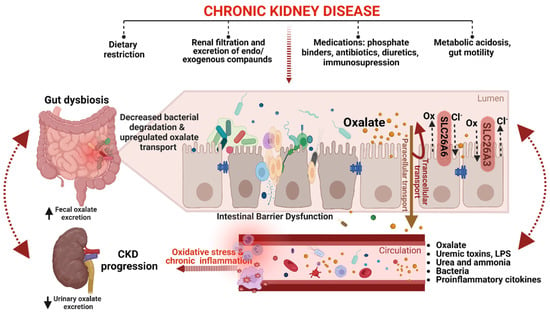
Figure 23. A schematic representation of the gut–kidney axis in CKD oxalate homeostasis (created with BioRender.com). CKD-associated changes in the gut microbiota can modify the abundance of ODB, leading to decreased oxalate degradation in the gut and resulting in hyperoxalemia. Additionally, CKD can affect the expression and activity of transporters, such as SLC26A6, which are involved in oxalate absorption and secretion in the gut, leading to increased fecal oxalate excretion as a compensatory mechanism to maintain oxalate homeostasis. Intestinal barrier dysfunction, on the other hand, can cause the translocation of uremic toxins, proinflammatory cytokines, bacteria, and their byproducts to the circulation, triggering oxidative stress and chronic inflammation. Furthermore, intestinal barrier dysfunction can also affect enteric oxalate handling, thereby exacerbating oxalate-mediated CKD progression. Dietary restriction, medications, gut motility, and metabolic acidosis are some of the factors that can also affect both intestinal barrier dysfunction and oxalate handling in CKD. However, the crosstalk between intestinal barrier dysfunction and enteric oxalate handling in CKD should be an area of future research.
5. Oxalate as a Clinical Marker for CKD Progression and Prognosis
Increased oxalate concentrations in both plasma and urine are commonly observed but often overlooked complications in patients with CKD. Hyperoxaluria (≥45 mg/24 h) is estimated to affect 5–24% of patients with gastrointestinal disease associated with malabsorption [92][109]. Although it is unclear how common hyperoxaluria is in CKD, it could have a significant impact, affecting approximately 250,000 individuals with gastrointestinal disease in the United States in 2019, which may potentially lead to kidney failure [93][110]. The prevalence of hyperoxalemia in CKD patients also remains unclear and has only recently been the subject of a few studies examining POx levels in both the general CKD population [30][94][95][57,111,112] and the dialysis cohort [96][113]. At reference values of 1–3 µmol/L, POx concentrations in patients with CKD range from 1 to ≥110 μmol/L, depending on estimated GFR (eGFR) [94][95][96][111,112,113]. These studies have shown a progressive increase in POx concentrations with decreasing eGFR, with the highest levels observed in patients undergoing hemodialysis (HD) [94][95][96][111,112,113]. However, despite the clear trend linking oxalate concentrations to eGFR in CKD patients, significant variations in POx and UOx levels have been reported among patients with the same eGFR level [30][95][96][57,112,113]. This issue is particularly exacerbated in anuric patients undergoing HD. Although the patient population is nearly homogeneous and underwent a standardized dialysis regimen, intraindividual POx concentrations vary significantly [82][96][97][102,113,114]. These variations can be attributed to other factors, such as differences in the amount of ingested oxalate, hepatic metabolism and intestinal absorption. For example, the daily UOx excretion increases by 2.7 mg per 100 mg of dietary oxalate ingested [98][6]. Patients with diabetes have been found to have increased plasma glyoxylate levels, which are intermediate molecules in the metabolic pathway of sugar and oxalate, resulting in increased POx and UOx concentrations [14][41]. Similarly, gastrointestinal disorders or medication intake may also affect bacterial ODA and oxalate absorption in the gut, determining POx and UOx concentrations in patients with ESKD and anuria [50][76][82][18,26,102].
Although there is ample in vitro and experimental evidence that oxalate plays a role in the development and progression of CKD, few clinical studies have examined the association between plasma or urinary oxalate levels and CKD progression. In a prospective cohort study, Wailkar et al. investigated the association between 24-h UOx excretion and the risk of CKD progression and kidney failure in 3123 participants with grades 2 to 4 CKD [99][115]. The study found that UOx excretion higher than 27.8 mg/24 h was independently associated with a 32% increased risk of CKD progression and a 37% increased risk of kidney failure [99][115]. Likewise, in a prospective study involving 167 stable kidney transplant recipients, a POx concentration greater than 13 µmol/L, measured over 10 weeks following transplantation, was found to be significantly associated with impaired long-term patient and graft survival during a 15-year follow-up period [36][63].
In addition to CKD progression, oxalate has also been linked to cardiovascular complications associated with CKD. Because oxidative stress and chronic inflammation are major risk factors for accelerated atherosclerosis [100][101][116,117], they seem to be the pathophysiological link between hyperoxalemia and cardiovascular disease (CVD) [63][87]. CaOx crystal deposits were observed in coronary arteriosclerotic lesions in patients treated with HD and peritoneal dialysis (PD) [102][118]. Elevated POx levels were also significantly associated with an increasing trend in atherogenic lipoprotein fractions and proinflammatory markers in patients undergoing dialysis kidney replacement therapy (DKRT) [31][58]. Moreover, in a prospective observation of 50 patients with ESKD, POx concentration ≥62.9 μmol/L was significantly associated with CVD events during the 2-year follow-up period, independent of other CVD risk factors [31][58]. Pfau et al. conducted a study in a well-established cohort of patients with type 2 diabetes mellitus undergoing HD, demonstrating a nearly linear increase in the risk of sudden cardiac death per doubling of POx concentration, where a POx concentration ≥59.7 µM was associated with a 40% increase in the risk of cardiovascular events and a 62% increase in the risk of sudden cardiac death, compared to those with a POx concentration ≤29.6 µM [32][59].
Despite compelling evidence suggesting the involvement of oxalate in CKD development and progression and its potential as a clinical marker, several limitations currently preclude oxalate from being used as a reliable clinical marker for CKD progression. One major limitation is the lack of standardized methods for measuring oxalate concentrations in clinical samples, leading to variability and inconsistency in reported results [30][57]. Additionally, oxalate levels can be influenced by various factors, such as diet, gut microbiota, and medication use, which can confound the interpretation of results. Furthermore, most of the existing studies on oxalate and CKD have focused on urolithiasis or primary hyperoxaluria populations, which may not accurately reflect the oxalate level and its clinical associations in the broader CKD population. Therefore, while oxalate may have potential as a clinical marker for CKD progression, further research is needed to establish standardized methods for measuring oxalate levels and to determine its role in a broader population of non-stone-forming patients with CKD.
6. Targeting Oxalate Homeostasis to Reduce CKD Progression and the Risk of Cardiovascular Events
Despite the increasing awareness of oxalate’s involvement in CKD pathogenesis and its correlation with cardiovascular outcomes, there are currently no approved treatments that specifically target mitigating oxalate burden in CKD patients. This creates a challenge for patients and healthcare providers in managing oxalate-related issues in the context of CKD, especially for those undergoing DKRT. Managing oxalate balance to prevent CKD progression necessitates a multifaceted approach, but only limited treatment options are currently available.