From bacteria to plants and humans, the glutathione system plays a pleiotropic role in cell defense against metabolic, oxidative and metal stresses. Glutathione (GSH), the γ-L-glutamyl-L-cysteinyl-glycine nucleophile tri-peptide, is the central player of this system that acts in redox homeostasis, detoxification and iron metabolism in most living organisms. GSH directly scavenges diverse reactive oxygen species (ROS), such as singlet oxygen, superoxide anion, hydrogen peroxide, hydroxyl radical, nitric oxide and carbon radicals. It also serves as a cofactor for various enzymes, such as glutaredoxins (Grxs), glutathione peroxidases (Gpxs), glutathione reductase (GR) and glutathione-S-transferases (GSTs), which play crucial roles in cell detoxication. This review summarizes what is known concerning the GSH-system, from cyanobacteria the ancient prokaryotes which have evolved it, to higher eukaryotes.
1. Introduction
Most life forms are continuously challenged with toxic
reactive
oxygen
species (ROS) present in our oxygenic atmosphere (ozone, O
3), and/or generated by respiration and cell metabolism
[1][2][3][1,2,3] and photosynthesis in cyanobacteria
[4][5][6][7][8][4,5,6,7,8], algae and plants
[9][10][11][9,10,11]. In addition, photosynthetic organisms are exposed to solar UV that also generate ROS
[12][13][12,13].
ROS molecules encompass singlet oxygens (
1O
2), superoxide anions (O
2●−), hydrogen peroxides (H
2O
2), and hydroxyl radicals (
●OH) that cause damages to target molecules, namely: lipids, nucleic acids and proteins
[2][10][2,10], thereby generating cell death in microorganisms and multiple disorders and diseases in humans
[14][15][16][14,15,16] that reduce longevity
[17].
Superoxide anions and hydrogen peroxides can both react with proteins containing iron-sulfur [Fe-S] clusters, liberating their Fe ions. Free or complexed Fe
2+ ions reduce H
2O
2, yielding hydroxyl radicals that modify all kinds of biomolecules at a diffusion-limited rate. Hence, radicals, sulfenic acids, disulfides and (hydro)peroxides are directly or indirectly formed by ROS
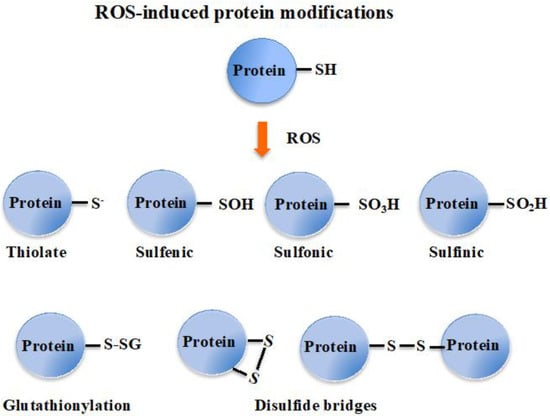
[3][18][3,18]. ROS also oxidize cysteines to form thiyl (sulfenyl) radical (-S
●) by one-electron transition; sulfenic acid (-SOH) and disulfide (-S-S-) by a two-electrons transition; sulfinic acid (-SO
2H) by a four-electrons transition; and eventually sulfonic acid (-SO
3H) by a six-electrons transition
[19]. Concerning disulfides, two types can be distinguished considering whether they link two cysteinyl residues from either the same or different proteins (intra- or inter-molecular disulfide bridges), or from a protein and a molecule of glutathione (glutathionylation). Glutathione is the γ-L-glutamyl-L-cysteinyl-glycine tri-peptide (hereafter designated as GSH) that plays a prominent role in ROS detoxification from bacteria to higher eukaryotes
[3][11][16][20][21][3,11,16,20,21]. It directly scavenges ROS and also serves as a redox cofactor for various antioxidant enzymes, such as glutaredoxins (Grxs), glutathione peroxidases (Gpxs), glutathione reductase (GR) and glutathione S-transferases (GSTs). The above-mentioned glutathionylation can protect cysteinyl residues against irreversible oxidation (generation of sulfinic and sulfonic acids), and/or act in regulation
[9][11][18][22][9,11,18,22], as shown in
Figure 1.
Figure 1. Schematic representation of the oxidation of the cysteinyl residue of protein to sulfenic (-SOH), sulfinic (-SO2H) and sulfonic (-SO3H); and disulfide (-S-S-) with another cysteinyl residue from the same or another protein, or a molecule of glutathione.
ROS can also be detoxified by various metabolites (ascorbate, carotenoids, vitamins, etc.) and several enzymes
[23]. The superoxide dismutase (SOD) converts O
2●− to H
2O
2, which is then detoxified to H
2O by the catalase and peroxidase enzymes
[18]. H
2O
2 can also be detoxified by the hydroperoxide activity of some glutaredoxins
[18]. The protein disulfides and glutathione-protein mixed disulfides are repaired by thioredoxins
[20][24][20,24], glutaredoxins
[3][11][25][26][3,11,25,26] and glutathione-S-transferases
[14][16][25][27][28][29][14,16,25,27,28,29].
ROS-removing systems are usually viewed as beneficial antioxidants that maintain damaging ROS below dangerous levels
[3][20][30][3,20,30]. However, ROS are also a necessary part of subcellular and intercellular communication in living organisms
[3][18][3,18]. Indeed ROS species can serve as signal mediators in the redox regulation of cell metabolism
[19], as they are enzymatically produced and degraded by NADPH-oxidases, which generate superoxide anions
[3], SOD, which generates H
2O
2, and catalase and peroxidase, which detoxify H
2O
2 into H
2O. Furthermore, H
2O
2 oxidizes protein thiols in disulfides or sulfenic acids, which can be reduced back to thiols, and are thereby good thiol redox switches for signaling
[10][18][10,18].
2. Biological Importance and Biotechnological Interests of Cyanobacteria
Cyanobacteria are primordial prokaryotes regarded as the “inventor” of oxygenic photosynthesis
[31][32], which played an important role in the evolution of Early Earth and the biosphere by absorbing a huge amount of the greenhouse gas carbon dioxide (CO
2), and evolving a huge amount of dioxygen (O
2)
[32][33][34][35][36][33,34,35,36,37]. Indeed, cyanobacteria are regarded as responsible for the oxygenation (and oxidation) of the atmosphere since the Great Oxidation Event around 2.4 Ga
[31][32][37][38][39][40][41][32,33,38,39,40,41,42].
As a consequence, cyanobacteria have long been challenged by ROS
1O
2, O
2●−, H
2O
2 and OH
[2][3][18][2,3,18], which are generated by their active photosynthesis and, sometimes, their respiration
[8][10][11][8,10,11]. Singlet oxygens are unavoidably produced by the interaction of sunlight with photosynthetic pigments (chlorophyll a, carotenoids and phycobiliproteins) while superoxide anions, hydrogen peroxides and hydroxyl radicals are generated when the light-driven electron transport exceeds what is needed for nutrients assimilation
[8][10][42][8,10,43]. Cyanobacteria are also strongly exposed to solar UV radiations (UVR) that also generate ROS
[20][43][20,44]. Consequently, cyanobacteria represent a major source of ROS in aquatic environments
[44][45].
Cyanobacteria are also frequently challenged by heavy metals (cadmium, cesium, chromate, mercury, lead, uranium, etc.) which are released by natural sources (volcanoes and forest fires) and anthropogenic activities (mining, fossil fuel burning, etc.). The toxicity of heavy metals is based on their chemical properties, which allow them to promote the production of ROS and the inactivation of enzymes
[45][46][47][47,48,49], basically by reaction with SH groups, including that of GSH
[48][49][50,51]. The presence of heavy metals in soils and waters is especially problematic because metals are persistent in the environment and they accumulate throughout the food chain, thereby threatening human health
[50][51][52][46,52,53]. Cyanobacteria are important organisms to investigate the relations between metals and oxidative stress as they constitute the first biological barrier against entry of heavy metals into the food chain.
To cope with ROS and environmental stresses, cyanobacteria have evolved the glutathione system
[5][53][54][5,59,60], which is crucial to their photoautotrophic lifestyle
[4][43][55][4,44,61] and has been conserved during evolution
[5][6][53][54][56][5,6,59,60,62]. The glutathione system comprises the glutathione tripeptide itself (γ-L-glutamyl-L-cysteinyl-glycine, GSH) and its cysteine-less homologs (ophthalmate and norophthalmate); in humans, these serve as biomarkers of diseases (See below), as well as numerous GSH-dependent enzymes, such as glutaredoxins and glutathione-S-transferases
[18][57][18,63], which have been conserved during evolution
[11][26][27][56][58][59][11,26,27,62,64,65]. In addition, cyanobacteria possess other promiscuous antioxidant enzymes, such as superoxide dismutases, catalases and peroxidases
[8].
Contemporary cyanobacteria continue to play a key role in the global ecosystem. They fix enormous amounts of atmospheric CO
2 and N
2 to produce huge amounts of O
2 and biomass for our food chain
[31][32][35][36][60][61][62][63][64][32,33,36,37,56,66,67,68,69]. They have been consumed by humans and used as soil fertilizers for over a thousand years
[65][66][67][68][70,71,72,73]. Furthermore, they produce a wealth of metabolites, such as vitamins, antioxidants (such as ergothioneine mentioned below), antibiotics, antifreezes, drugs, osmoprotectants and toxins
[60][69][70][71][72][56,74,75,76,77] that can influence human health and/or improve plant growth and/or resistance to stress (drought, salt, heavy metals and pathogens)
[73][78].
Finally, in colonizing most aquatic ecosystems and soils of our planet, where they face various environmental challenges and interactions with competitors, predators or symbiotic hosts (angiosperms, bryophytes, fungi and gymnosperms), cyanobacteria have evolved as widely diverse organisms. They display various cell morphologies
[74][83] and cellular differentiation
[75][84], as well as widely diverse genome sizes (1.44–12.07 Mb), GC content (30–60%) and organization (presence of a circular chromosome with or without one to several linear chromosomes and circular plasmids)
[76][77][82,85].
3. Synthesis and Importance of Glutathione in Living Organisms
Glutathione (GSH) was discovered in 1888 by J. de Rey-Pailhade, and its composition as γ-L-glutamyl-L-cysteinyl-glycine was established much later, in 1935
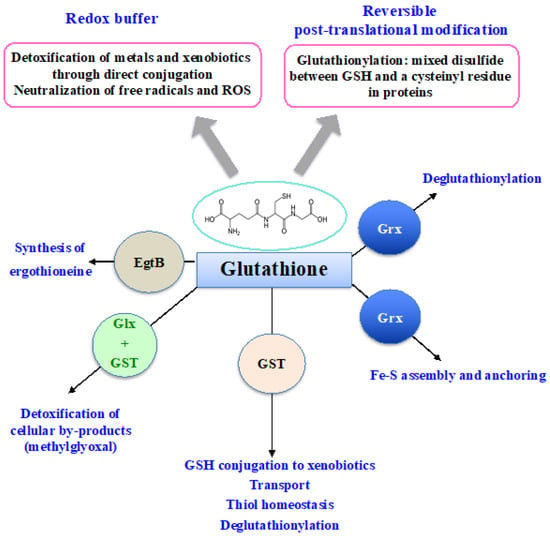
[78][86]. That the Cys and Glu of GSH are linked through the γ-carbonyl group of Glu instead of the typical α-carboxyl group confers a high stability to GSH since only very specific enzymes under particular conditions may operate on its degradation (See below). GSH is the most abundant non-protein thiol (concentration ranging from 0.1 mM to about 20 mM) in all three kingdoms of life: Bacteria (mostly Gram-negative, rarely Gram-positive), Archaea, and Eukarya, where it plays pleiotropic roles in cell life and resistance to stresses
[3][5][11][15][18][20][21][79][80][81][3,5,11,15,18,20,21,87,88,89]. GSH is a nucleophilic metabolite that directly scavenges ROS, nitric oxides and carbon radicals
[3][5][11][18][3,5,11,18]. GSH also serves as electron donor to various antioxidant enzymes, including glutaredoxins, glutathione peroxidases and glutathione-S-transferases (See below). Furthermore, GSH can also act in the synthesis of ergothioneine, another antioxidant catalyzed by the EgtB enzyme (See below). In addition, the cysteinyl thiols of GSH can complex metal
[3][15][20][3,15,20], as shown in
Figure 23.
Figure 23.
Schematic representation of the pleiotropic roles of glutathione.
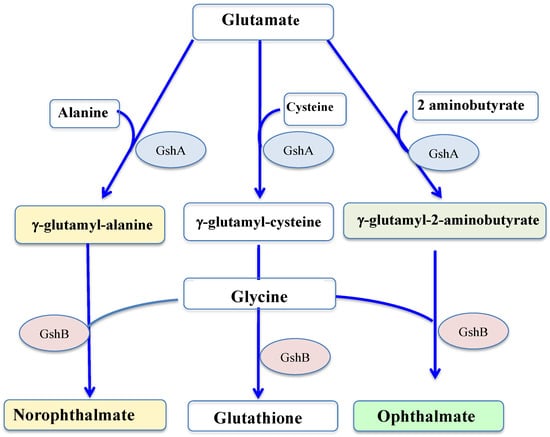
GSH is synthesized by two sequential ATP-requiring enzymes, namely the γ-glutamyl-cysteine (γ-GC) synthetase (γ-GCS, EC 6.3.2.2), which forms γ-GC from L-glutamic acid and L-cysteine, and the GSH synthetase (GS, EC 6.3.2.3), which forms GSH from γ-GC and L-glycine, as shown in
Figure 34.
Figure 34. Schematic representation of the synthesis of glutathione and its thiol-less analogs. GshA: gamma-glutamylcysteine ligase; GshB: glutathione synthase.
Glutathione is vital in many organisms, including yeast
[20][82][83][20,94,95], mice
[84][96], plants
[10][79][85][10,87,97] and cyanobacteria
[4][55][4,61], but not in
Escherichia coli [20][86][87][88][20,92,98,99].
E. coli mutants devoid of GSH do not exhibit enhanced sensitivity to oxidative stress (H
2O
2, cumene hydroperoxide, ionizing (gamma) radiations) in exponentially growing culture
[86][92], but stationary-phase cultures are more sensitive to H
2O
2 than the wild-type
[89][100].
In plants, where the complete absence of GSH causes death at the embryonic stage
[85][97], mutants with a less severe decrease in GSH content are viable but more sensitive to many biotic and abiotic stresses
[11], including Cd
[90][105] and Zn
[91][106]. The biosynthesis of γ-glutamyl-cysteine catalyzed by glutamate cysteine ligase takes place in chloroplasts
[9], which likely originated from cyanobacteria
[33][92][93][34,107,108], while the formation of GSH catalyzed by glutathione synthetase can occur both in the chloroplasts and in the cytosol. Then, GSH is transported to mitochondria and the nucleus
[9][11][18][94][9,11,18,109].
Some Gram-positive bacteria, including
Actinobacillus pleuropneumoniae [95][122],
Listeria monocytogenes [96][123],
Pasteurella multocida [97][124] and both
Streptococcus agalactiae and
Streptococcus thermophilus [98][99][125,126], contain a newly discovered bifunctional enzyme, termed GshF, which possesses both GshA and GshB activities. The N-terminal sequence of GshF is similar to that of
E. coli GshA, but the C-terminal sequence is more similar to the D-Ala, D-Ala ligase than to any known GshB
[98][125].
Due to its critical role in antioxidation, xenobiotic detoxification, and immune regulation pathways, GSH has been widely used in the food, cosmetic, and pharmaceutical industries
[100][131]. So far, GSH is commercially produced mainly by
Saccharomyces cerevisiae strains, which have generally been recognized as safe. Previous studies have focused on overproducing GshA and GshB, but the production yield and titer of GSH in such
Saccharomyces cerevisiae strains remain low due to the feedback inhibition on GshA
[101][127]. To overcome this limitation, the GshF bifunctional enzyme from Gram-positive bacteria was produced in
S. cerevisiae, as GshF is insensitive to feedback inhibition. The resulting strain produced 240 mg L
−1 GSH with GSH content and yield of 4.3% and 25.6 mg
glutathione/g
glucose, respectively
[101][127]. However, this production of GSH by
S. cerevisiae competes with glucose demands of other industries and results in high production costs
[100][131].
Instead of GSH itself, some organisms employ its precursors or derivates
[3][53][102][3,59,120], such as γ-glutamyl-cysteine in halobacteria and halophilic archaea or trypanothione in kinetoplastid parasites
[103][132]. They may also use other thiols, e.g., bacillithiol in Gram-positive Firmicutes
[104][133] or mycothiol in many actinobacteria, such as the human pathogen
Mycobacterium tuberculosis [105][134].
In some organisms, such as mycobacteria
[106][135], the γ-glutamyl-cysteine peptide is also used for the synthesis of ergothioneine (hereafter EGT), an unusual thio-histidine betaine amino acid (also known as 2-mercaptohistidine trimethylbetaine) that has potent antioxidant and cytoprotective activities
[107][108][109][110][111][136,137,138,139,140]. Hence, in mycobacteria there is competition between EGT and glutathione biosynthesis. EGT has both a thiol (antioxidant) and a thione form
[107][111][136,140], with the latter thione tautomer being predominant at physiological pH, thereby making EGT unusually resistant to oxidation by molecular O
2 [108][137]. Its midpoint potential, +0.06 V, is unusually high compared to typical thiols, including GST (−0.2 to −0.4 V)
[108][137]. EGT can serve as a reductant via one-electron reaction or as a nucleophilic reagent via two-electrons exchange.
Few other organisms, such as cyanobacteria (see below) and certain fungi (
Neurospora crassa, the fission yeast and mushroom fruiting bodies) are able to synthesize EGT
[107][112][113][136,141,142], unlike plants and animals who acquire it via their soil and their diet, respectively
[106][108][135,137]. EGT is stable in the body for a long time after ingestion, and is viewed as protecting the central nervous system against diseases
[109][138]. Animals have evolved a highly selective transporter for it, originally known as a carnitine transporter (OCTN1)
[109][111][138,140] and also called ergothioneine transporter ETT because of its 100-fold higher affinity for EGT
[110][139]. Genetic analysis in mycobacteria
[113][142] and EGT consumption in mammals has shown that EGT protects cells against oxidative
[108][110][137,139], metal
[114][143] and UV
[108][110][137,139] stresses.
Returning to glutathione, some of its homologs have no cysteine residue, and therefore no reducing properties, such as ophthalmate (L-γ-glutamyl-L-α-aminobutyryl-L-glycine) and norophthalmate (L-γ-glutamyl-L-alanyl-L-glycine). Ophthalmate (hereafter OPH) and norophthalmate (NOPH) were initially discovered in various animal organs (lens, brain and liver)
[115][116][144,145], where they are regarded as biomarkers of GSH depletion elicited by oxidative stress
[117][118][146,147]. OPH was also found to have accumulated in stressed plants
[119][148], yeasts
[120][149], bacteria (
E. coli)
[121][150] and cyanobacteria (
Synechocystis PCC 6803)
[55][61].
4. Glutathione Degradation
Because of its γ-linkage between the carboxyl group of glutamate and the amine group of cysteine, GSH cannot be degraded by genuine protease. Dedicated peptidases, γ-glutamyl transpeptidase (GGTs; E.C. 2.3.2.2) and/or γ-glutamyl cyclotransferases (γ-GCT or GGCTs, EC 4.3.2.9), catabolize GSH in bacteria and/or eukaryotes
[9][11][21][122][9,11,21,110].
The GGT enzyme is conserved throughout all three domains of life
[11][16][123][11,16,173]. Bacterial GGTs are generally soluble and localized in the periplasmic space or secreted in the extracellular environment
[123][173], whereas eukaryotic GGTs are embedded in plasma or vacuole membranes
[9]. GGT enzymes release the cysteinyl-glycine dipeptide and 5-oxoproline, a cyclized form of glutamate (Zhang and Forman, 2012). Then, the cysteinyl-glycine dipeptide is broken down into cysteine and glycine by specific Cys-Gly peptidases (including the leucine aminopeptidase (EC 3.4.13.18)) while the 5-oxoproline is converted into glutamate by the ATP-dependent 5-oxoprolinase (EC 3.5.2.9). The released glutamate, cysteine and glycine can be ploughed back into the synthesis of reduced GSH
[9][11][9,11]. In
E. coli and a few other bacteria, the tripeptidase (PepT) also acts in GSH degradation, and both the GGT- and the PepT-encoding genes are dispensable for cell growth under favorable laboratory conditions
[9][21][9,21].
In plants, GSH degradation seems to be as important as GSH synthesis for sulfur metabolism
[122][110]. The apoplastic and vacuolar GSH pools are degraded by GGTs, which cleave the γ-glutamyl moiety of GSH, GSSG and GS-conjugates, or transfer the Glu residue is transferred to amino acids to produce γ-Glu amino acids
[9][11][122][9,11,110], which are beneficial for human consumption
[124][164].
Arabidopsis possesses four GGTs. GGT1 and GGT2 are localized in the apoplast and degrade extracellular GSSG into Glu and Cys–Gly
[122][110], similar to mammalian GGTs. The Cys–Gly dipeptide is further broken down by a dipeptidase into Cys and Gly. They are then translocated to the cytosol where they serve in the synthesis of protein and GSH, followed by a novel round of export/degradation in the apoplast
[9].
GGT3 is considered a pseudogene since its transcript encodes a protein lacking a sequence important for GGT catalytic activity. The
GGT4-encoded isoform is present in the vacuole where it supports detoxification processes by degrading GSH conjugates formed by glutathione S-transferase following exposure to toxic xenobiotics
[11][28][122][11,28,110]. GSH degradation mainly occurs in the cytosol
[11].
In mammals, where it was first reported, the GGT enzyme is a cell-surface protein that contributes to the extracellular catabolism of GSH, but it has no role in either GSH or γ-glutamyl-Cys transport back into cells
[9]. Interestingly, GGT is used as a diagnostic marker for many human diseases
[16][125][16,174]. Mammals also have both ChaC1- and ChaC2-type GGCT enzymes, which are expressed only under endoplasmic reticulum stress or constitutively, respectively. ChaC1 and ChaC2 have high specificity and comparable
Km values for GSH, but ChaC1 has 10- to 20-fold higher catalytic activity than ChaC2
[122][110].
5. Glutathione Reductase
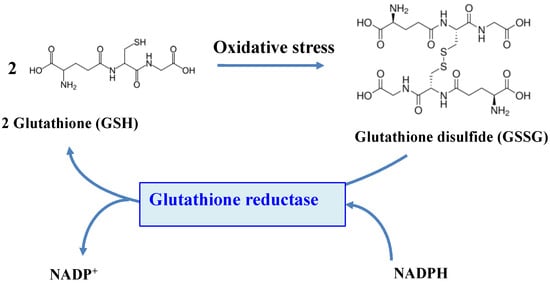
The function of GSH depends on the reactivity of its cysteinyl thiol group, which can complex metals, be alkylated to thioethers or oxidized to disulfides, thereby forming a glutathione disulfide dimer (GSSG)
[3][15][20][3,15,20]. Four processes can remove GSSG after oxidative challenge: (i) ATP-driven export of GSSG
[20]; (ii) degradation of GSSG by peptidase
[9][11][9,11]; (iii) reduction of GSSG by the glutathione reductase (GR) enzyme
(see belo
w and Figure 2) or the thioredoxin reductase/thioredoxin (TrxR/Trx) couple
[3][15][20][3,15,20] and (iv) the glutathionylation/deglutathionylation of proteins yielding RSSG and GSH
[11][18][22][11,18,22]. Under normal conditions, GSH is about a 100-fold more abundant than GSSG
[18][20][21][18,20,21]. For example, the GSH/GSSG molecular ratio is about 200 in
E. coli cells growing in the rich standard-medium LB.
The flavoenzyme GR (EC 1.8.1.7) is a highly conserved enzyme across the tree of life, which converts GSSG to two molecules of GSH, by using NADPH (mostly) or NADH (rarely) as the reducing agents
[11][15][18][20][21][126][127][128][129][11,15,18,20,21,175,176,177,178], as shown in
Figure 45.
Figure 45.
Schematic representation of the role of the glutathione reductase enzyme.
GR belongs to the pyridine nucleotide-disulfide oxidoreductase family that includes the related enzymes dihydrolipoamide dehydrogenase, mercuric ion reductase trypanothione reductase and some TrxR-isoforms
[3]. GR from pro- and eukaryotes share about 50% identity and form stable homodimers of ~110 kDa each comprising three domains
[3][102][126][3,120,175]. The FAD- and NADP-(binding) domains are globular, whereas the interface dimerization domain is somewhat flat. It contains two regions, at the N-terminus 71–104 and C-terminus 372–482. The FAD-binding and the NADPH-binding domains are in residues 1–157 and 198–238, respectively. The catalytic site of GRs possesses two conserved cysteines (C61, C65) that can form a disulfide bond. In the
S. cerevisiae enzyme, the cysteine C239 (not conserved) can bind excess GSH when required.
GR accumulate in cellular regions of high electron flux, where ROS are generated
[102][120]. In prokaryotes, GR is localized in the periplasm, associated with the inner membrane facing the cytoplasm, and it can be secreted to the extracellular environment. In eukaryotes, GR is present in the cytoplasm, the endoplasmic reticulum lumen, the lysosomes and the organelles nucleus, mitochondria and chloroplasts, thanks to its transport from the cytosol
[3][11][18][20][102][126][129][3,11,18,20,120,175,178].
In
E. coli, GR deficient mutants, originally isolated in a screen for diamide sensitive mutants
[130][131][180,181], have wild-type (WT) growth rates under standard conditions
[20]. They are more sensitive than the WT strain to cumene hydroperoxide and the O
2●−-generating compound paraquat, but not to
t-butyl hydroperoxide
[132][133][182,183]. Furthermore, their increased sensitivity to H
2O
2 could be uncovered only in a catalase mutant background
[134][184]. Despite their weak GR activity (0.45 units as compared to 35 units in WT cells), the ratio of GSH to GSSG is not altered significantly from that of WT. This finding indicates that GSSG can also be reduced by other enzymes, such as thioredoxin reductase/thioredoxin (TrxR/Trx) couples
[3][20][131][3,20,181].
In
Saccharomyces cerevisiae and human cells, the GR-encoding gene was cloned on the basis of its sequence homology with the
E. coli gene
[20]. This eukaryotic GR gene expresses both the cytosolic and mitochondrial forms of GR, which are synthesized using alternative in-frame start codons. Starting at the first AUG codon, the synthesis generates a long GR isoform marked for transport to the mitochondria. The translation starting at the second AUG codon generates a shorter GR isoform remaining in the cytosol. Usually, the pre-sequence of the mitochondrial form is cleaved off upon import by mitochondrial proteases so that the mitochondrial and cytosolic forms have a similar length
[20][102][20,120]. Yeast mutants lacking GR show WT growth, but accumulate increased levels of GSSG
[135][136][185,186] and increased export of GSSG into the vacuole to maintain the highly reducing environment of the cytosol
[18]. These mutants are very sensitive to H
2O
2 and the thiol oxidant diamide and are partially sensitive to cumene hydroperoxide, t-butyl hydroperoxide and paraquat
[20][137][20,187].
In humans, GR activity is positively correlated to longevity, and centenarians have an increased level of GR
[17], but cancer cells in having high levels of GSH and GR are refractory to some therapies that induce oxidative stress
[102][120]. Low GR activity is correlated with a higher susceptibility of cataract development during early adulthood
[3] and HIV-1 infection
[138][118]. In mice, GR was shown to act in the defense against bacterial infections
[139][188].
In plants, GR activity is increased in response to abiotic stresses triggered by heavy metals, salts, drought, UV radiation and chilling temperatures
[10][11][10,11]. Transgenic approaches elevating GR activity increased resistance to oxidative stress, whereas mutants with lower GR activities were more sensitive to oxidative stress and were affected in their development
[3][10][3,10].
6. Importance of the Evolutionary-Conserved Glutathione-S-Transferase Enzymes
As the reactivity of glutathione (GSH) with proteins, small molecules and xenobiotics can be low in vivo
[18][140][18,205], GSH-dependent reactions are accelerated by enzymes, such as glutaredoxins (See below) and glutathione-S-transferases (GSTs).
Glutathione-S-transferases (EC 2.5.1.18) constitute a superfamily of enzymes that play prominent roles in specialized secondary metabolism and detoxication. The presence of GSTs in most living organisms highlights their ancient origin and the preservation of their functions during evolution
[3]. GSTs are widely studied in eukaryotes because of their great relevance to human health
[14][58][141][142][143][144][14,64,206,207,208,209], plant growth
[28][145][28,210] and responses to stresses
[27][57][146][147][27,63,211,212] and pathogens
[148][213]. GSTs are less well studied in prokaryotes even though they act in bacterial protection against metabolite by-product (methylglyoxal, see below)
[149][214] and pollutants such as polychlorinated biphenyls (PCBs), dichloroacetate and polycyclic aromatic hydrocarbons (PHA)
[150][151][152][215,216,217].
GSTs catalyze reactions where GSH is consumed (GSH-conjugation on metabolites, chemicals or metals) or not (isomerization and dehalogenation) and reactions where GSH is oxidized (GSH-dependent peroxidases, -thiol-transferase, -dehydro-ascorbate reductase)
[3][18][3,18]. GSTs can also bind and transport molecules through their noncatalytic ligandin properties
[147][153][212,218].
GSTs are mainly homo- or heterodimeric enzymes, where each subunit contains an N-terminal thioredoxin (TRX) domain linked to an α-helical C-terminal domain
[14][145][154][14,210,219]. The active site, located in a cleft between both domains, contains a GSH-binding site and a hydrophobic-substrate binding site. Based on their amino-acid sequence, GSTs were classified into various classes designated by a Greek letter
[8][155][8,220]. GSTs having a sequence identity greater than 40% or lower than 25% belong to the same class or different classes, respectively
[150][215]. GSTs are also grouped based on their localization in the cell, namely cytosolic, mitochondrial and microsomal, commonly referred as membrane-associated proteins in eicosanoid and glutathione metabolism (MAPEG)
[3][18][56][150][152][154][3,18,62,215,217,219].
7. Glutathione Acts in the Detoxification of Methylglyoxal, a By-Product of Cell Metabolism from Cyanobacteria to Humans
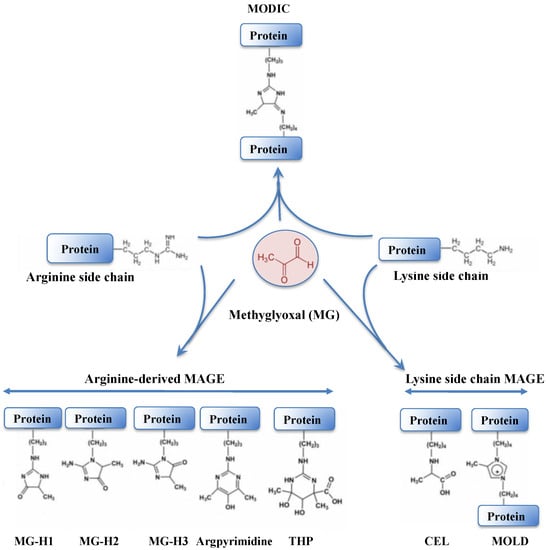
Methylglyoxal (MG) is a very dangerous dicarbonyl compound that strongly interacts with lipids, nucleic acids and the arginine and lysine residues of proteins (
Figure 57), generating advanced glycation end products (AGEs) that disturb cell metabolism in prokaryotes
[156][157][230,231] and eukaryotes
[156][158][159][230,232,233].
Figure 57. Schematic representation of protein modification resulting from the crosslinking of methylglyoxal onto arginine or lysine amino-acid residues. The abbreviations are as follows: MODIC, 2-ammonio-6-({2-[4-ammonio-5-oxido-5-oxopently)amino]-4-methyl-4,5-dihydro-1H-imidazol-5-ylidene}amino)hexanoate; MG-H1, N(delta)-(5-methyl-4-imidazolon-2-yl)-L-ornithine; MG-H2, 2-amino-5-(2-amino-5-hydro-5-methyl-4-imidazolon-1-yl)pentanoic acid; MG-H3, 2-amino-5-(2-amino-4-hydro-4-methyl-5-imidazolon-1-yl)pentanoic acid; THP, N(delta)-(4-carboxy-4,6-dimethyl-5,6-dihydroxy-1,4,5,6-tetrahydropyrimidine-2-yl)-L-ornithine; CEL,
Nε-(carboxyethyl)lysine and MOLD: 1,3-di(
Nε-lysino)-4-methyl-imidazolium.
Like ROS, MG has a dual nature depending on its concentrations within the cells; it acts in signaling at low concentrations, but provokes detrimental effects, such as glutathionylation
[160][234] (see below), at high concentrations
[158][161][232,235]. In humans, MG is implicated in diabetes
[162][236] and age-related disorders
[156][230], such as retinopathy, nephropathy, cancer, and Parkinson’s and Alzheimer’s diseases
[159][160][163][233,234,237]. Hence, MG is increasingly regarded as a marker of diabetes-related diseases. The calculated MG concentrations in mammalian cells were reported to vary between 0.5 and 5 μM, similar to what was observed in yeast (4 μM)
[18]. In plants, elevated MG levels are a general response to abiotic and biotic stresses, such as salinity, heavy metals and drought
[11]. Furthermore, MG is viewed as acting in signaling via abscisic acid, Ca
2+, K
+ and ROS, and these processes are regarded as useful for the development of stress-resilient crops
[11][158][164][11,232,238].
MG is mainly formed in all cells both under normal and pathological conditions by the non-enzymatic breakdown of the triose phosphate isomers dihydroxyacetone phosphate (DHAP) and glyceraldehyde-3-phosphate (G3P)
[156][157][159][160][230,231,233,234], which rapidly lose α-carbonyl protons and their phosphate groups, generating MG. MG is also generated by the spontaneous auto-oxidation of ketone bodies and sugars, lipid peroxidation and the Maillard reaction between reducing sugars and amino acids
[165][166][239,240].
MG is predominantly detoxified by the glyoxalase pathway, which starts by the supposedly “spontaneous” (non-enzymatic) conjugation of MG with GSH to form a hemithioacetal (HTA). HTA is then isomerized by glyoxalase I (GlxI,
S-d-lactoylglutathione lyase; EC 4.4.1.5) to S-d-lactoylglutathione that is hydrolyzed by glyoxalase II (GlxII,
S-2-hydroxyacylglutathione hydrolase; EC 3.1.2.6) to produce D-lactate and release GSH
[3][11][18][162][163][167][3,11,18,236,237,242]. That GSH is required for MG detoxication explains why MG is accumulated in response to GSH depletion caused by oxidative stress
[160][234]. GlxI belongs to vicinal oxygen chelate enzymes, which contain an ancient βαβββ-motif required for binding metal ions
[3][160][3,234]. GlxI (also called Glo1) from humans, yeast and the parasite
Plasmodium falciparum, is a dimeric protein that prefers Zn
2+ (or Fe
2+) at its active site, whereas the enzymes from
E. coli and the pathogens
Yersinia pestis,
Pseudomonas aeruginosa and
Neisseria meningitidis and the protist
Leishmania major are optimally activated in the presence of Ni
2+ (or Co
2+)
[163][237].
8. Glutathione Maintains the Redox Homeostasis of Protein Thiols via Glutathionylation/Deglutathionylation Catalyzed by Glutathione-S-Transferases and Some Glutaredoxins
Oxidative stress promotes the covalent modification of proteins by GSH, i.e., formation of a disulfide bridge between the thiol group of a cysteinyl residue of a protein and a molecule of GSH
[3][18][140][3,18,205]. This post-translational modification called S-glutathionylation is regarded as a transient protection of critical cysteines against irreversible oxidation towards sulfinic and sulfonic acid forms during oxidative stress
[25][168][25,247]. It occurs only at specific cysteinyl residues of proteins, in response to ROS and MG
[160][169][234,248], and not randomly. A basic environment or the proximity of a metal cation are key determinants for the tendency of thiol groups to become deprotonated and consequently be affected by oxidation and spontaneous S-glutathionylation
[11]. As GSH is a bulky molecule, its ligation to proteins can have an impact on their structure, function, catalytic capacity and/or subcellular localization
[11][22][25][29][142][11,22,25,29,207].
Grx are small thiol proteins found in all kingdoms of life
[3][10][18][22][26][29][170][3,10,18,22,26,29,253]. The first identified function of Grx was described as an electron donor for the ribonucleotide reductase enzyme (RNR) in a
E. coli mutant lacking Trx
[171][254], the classic hydrogen donor for RNR
[172][255]. Grx can detoxify hydroperoxide thanks to their hydroperoxidase activity
[18]. Bacterial Grxs have the most basic form of the Trx-fold, consisting of a four to five central β-sheet surrounded by three α-helices. Grxs of higher organisms frequently display additional N- and C-terminal helices. Interestingly, GSTs, the other glutathione-dependent enzymes, have similar architectures, supporting the theory of a common ancestor for Grxs and GSTs
[3][173][3,256].
Grx-isoforms can be structurally categorized as monomeric or dimeric proteins, which possess an active site with the sequence motif CXXC (dithiol Grxs) or CXXS (monothiol Grxs), with or without an Fe/S-cluster
[3][18][26][80][174][3,18,26,88,91]. Grx can be furthermore grouped based on enzymatic activities, subcellular localizations or (putative) physiological functions (ROS detoxication, iron metabolism, etc.)
[15][174][15,91]. In plants, Grx are involved in the regulation of development through interaction with distinct transcription regulators
[10]. In humans, Grx functions have been implied in various physiological and pathological conditions, from immune defense to neurodegeneration and cancer development
[26].
9. Glutathione, Glutaredoxins and the Biogenesis of the Iron-Sulfur Cluster of Proteins
Glutathione plays a crucial role in cellular iron metabolism
[15][175][176][177][178][15,90,266,267,268] and the synthesis and repair of iron-sulfur cluster [Fe-S] of a wealth of enzymes (See below). Iron (Fe) and sulfur (S) are crucial elements in all kingdoms of life
[177][178][179][267,268,269]. Iron, the fourth most plentiful element in the Earth’s crust
[180][270], is frequently a growth-limiting factor because ancient cyanobacteria raised the oxygen levels that oxidized the soluble ferrous ions (Fe
2+) to insoluble ferric ions (Fe
3+)
[32][34][37][38][40][181][182][33,35,38,39,41,271,272]. Fe atoms are associated with many proteins as part of hemes, mono- or di-iron non-heme centers, or iron–sulfur [Fe-S] clusters
[183][184][273,274]. Sulfur (S) is the fifteenth and the sixth most abundant chemical elements in Earth’s crust and aquatic environment, respectively
[180][270]. Sulfur is essential in living organisms and is notably required for the synthesis of cysteine, which also serves for the synthesis of GSH, [Fe-S] and methionine
[185][275]. Biological organisms absorb and assimilate sulfate from their environment via a reductive pathway involving a series of transporters, enzymes and GSH.
[Fe-S] clusters are critical cofactors in all categories of life. They participate in the transfer of electrons (photosynthesis and respiration), transcriptional and translation regulation, DNA repair and replication
[15][18][26][80][174][176][183][186][187][188][15,18,26,88,91,266,273,276,277,278]. The chemically simplest Fe-S clusters are the rhombic [2Fe-2S] and the cubane [4Fe-4S] types, which contain iron (Fe
2+/3+) and sulfide (S
2−)
[183][273]. [Fe-S] clusters were discovered in the early 1960s by purifying enzymes, including plant and bacterial ferredoxins, with characteristic electron paramagnetic resonance signals
[183][273]. Later, chemists and biochemists devised in vitro protocols to assemble [Fe-S] clusters into apoproteins, and thereby assumed that these co-factors can assemble spontaneously on proteins in living cells.
[2Fe-2S] centers are synthesized from iron and cysteine-derived sulfur by highly conserved multi-protein machineries, including the iron-sulfur cluster (ISC) synthesis machinery
[183][187][188][273,277,278] and many types of Grx
[15]. The first [Fe-S]-Grx were isolated from humans (CSYC-type Grx,
[189][279]) and the poplar tree (CGYC-type Grx,
[190][280]). In both enzymes, the apo form (monomer) is a regular redox active Grx, while the holo form (dimer) has a bridging [2Fe-2S] cluster but no oxidoreductase activity. This [2Fe-2S] center lies at the interface of a dimeric complex of two Grxs ligated by the two N-terminal active site thiols and the thiols of two non-covalently bound GSH molecules
[174][91].