Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 1 by Matteo Molica.
Gilteritinib is a next-generation tyrosine kinase inhibitor (TKI) primarily targeting FLT3FLT3 and AXL (an onco-genic tyrosine kinase) receptors.
- resistant/relapsed acute myeloid leukemia
- tyrosine kinase inhibitors
- gilteritinib
- miRNAs
- HSCT
1. Pharmacodynamics and Pharmacokinetics of Gilteritinib
Median maximum concentration is reached after 2–6 h following single and repeat dosing of oral gilteritinib (rapid absorption with or without food); mean elimination half-life was 113 h. Elimination was primarily via feces. Gilteritinib is primarily metabolized via cytochrome CYP3A4; coadministration of gilteritinib with itraconazole (a strong P-glycoprotein inhibitor and CYP3A4 inhibitor) or rifampicin (a strong P-glycoprotein inducer and CYP3A inducer) can significantly interfere with its pharmacokinetic profile [10]. Compared with first-generation multitargeted TKIs, it is more selective to FLT3FLT3 and has greater potency. It blocks FLT3FLT3 receptors’ ATP-binding site competitively, thus inhibiting receptor signaling and halting cell cycle [11]. Cellular experiments have shown powerful inhibitory effects on FLT3FLT3 mutations (FLT3FLT3–ITD and FLT3FLT3-D835Y point mutations in particular) [12]. Since both FLT3FLT3–ITD and FLT3FLT3–TKD mutations promote constitutive FLT3FLT3 kinase activity, sustaining leukemic cell proliferation and survival, gilteritinib-mediated inhibitory effects have the potential to lessen the leukemia burden of AML patients (Figure 1). It is classified as a type I inhibitor, generally unaffected by mutations in the activation loop (e.g., at D835) [13]. Moreover, gilteritinib promotes apoptosis in FLT3FLT3–ITD mutations carrying tumor cells in vitro [9]. In xenografted mice models, oral administration of gilteritinib lowered phosphorylated FLT3FLT3 levels by 40% after 1 h [12], while a single dosage was sufficient to reduce the phosphorylation of STAT-5, a known downstream FLT3FLT3 target [12]. Following successive 120 mg doses of gilteritinib in patients with R/R-AML, approximately 90% of FLT3FLT3 phosphorylation was decreased, with inhibition starting to take place 24 h after the first dosage [9]. When oral gilteritinib (1–10 mg/kg) was given to mice once every day for 28 days, tumor development was significantly suppressed by 63–100% (p = 0.05) [12]. Although gilteritinib did not influence the in vitro reduction in tumor growth or induction of apoptosis, stimulation of the FLT3 ligand can raise the chance of resistance to other FLT3 inhibitors [14]. Given that AXL activation is a known resistance mechanism to FLT3 inhibitors and that AXL inhibition can slow the growth of FLT3–ITD AML tumors, gilteritinib additional activity against AXL may also be advantageous [15]. In comparison with other less specific TKIs, gilteritinib may present a lower clinical risk of side events, such as myelosuppression [12]. Inhibition of c-KIT (an oncogene encoding KIT, a platelet-derived growth factor receptor essential for hematopoiesis) is expected to provoke severe myelosuppressive effects because FLT3 and KIT structures are remarkably similar [10]. Thus, the risk of myelosuppression with gilteritinib is anticipated to be lower than with other TKIs because it has no impact on c-KIT [10]. Based on in vitro findings, CYP3A4 primarily metabolizes gilteritinib [10]. The main metabolites identified in animal investigations are M17, M16, and M10 (all accounting for less than 10% of the parent exposure); it is unknown if these metabolites have any effect on FLT3 or AXL receptors [9]. Since gilteritinib is a P-glycoprotein (P-gp) substrate, a multidrug transporter that actively pumps substances out of the cell and away from their target regions [16], it might exert an inhibitory effect on BCRP, P-gp, and OCT1 in the small intestine as well as the liver [9]. In vivo, gilteritinib neither induces nor inhibits CYP3A4 or MATE1. Since gilteritinib may decrease the effectiveness of 5-HT2B or sigma nonspecific receptor targeting medications in vitro (such as escitalopram), it should only be used in rare conditions together with these medications [9]. Reduced gilteritinib plasma concentrations are caused by coadministration with a P-gp and potent CYP3A inducer, hence this should be avoided [9]. Conversely, gilteritinib exposure is increased when it is administered concurrently with a potent CYP3A and/or P-gp inhibitor [10]. For instance, coadministration of a single 10 mg dose of gilteritinib with 200 mg of itraconazole per day for 28 days raised Cmax and AUC in healthy individuals by 20% and 120%, respectively [9]. A concurrent strong CYP3A and/or P-gp inhibitor increased exposure in individuals with R/R-AML by about 1.5 times [9].
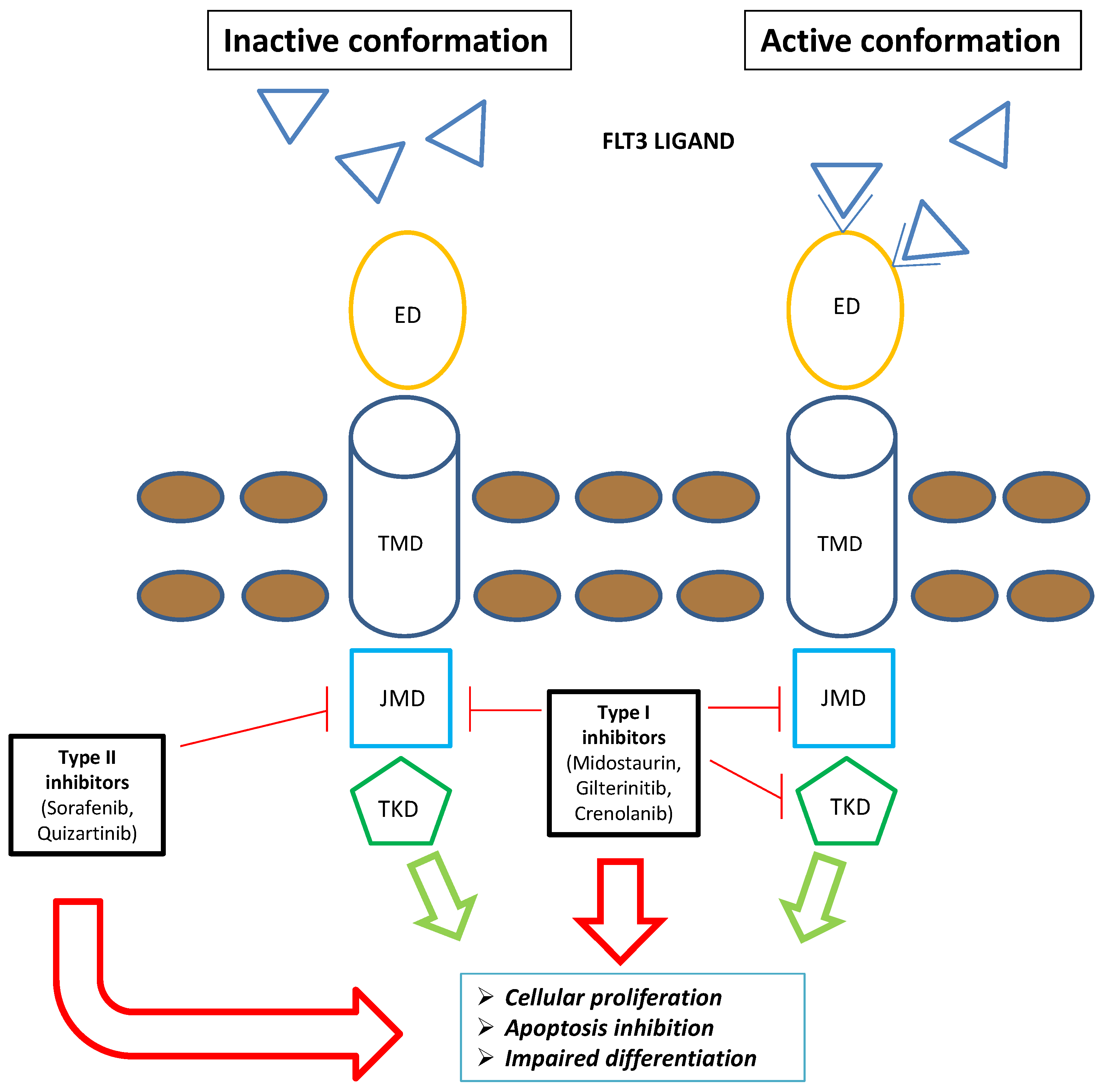
Figure 1. Schematic representation of FLT3 inhibitors’ mechanism of action: The type I family of FLT3 inhibitors (midostaurin, gilteritinib, and crenolanib) is able to bind the FLT3 receptor both in the active and inactive conformation, inhibiting FLT3–ITD and TKD mutations. Contrarywise, the type II family of FLT3 inhibitors (sorafenib and quizartinib) is able to bind the FLT3 receptor in the inactive conformation, acting only on FLT3–ITD. Overall, FLT3 inhibitors severely compromise leukemogenic activity of FLT3 (i.e., cellular proliferation, apoptosis inhibition, and impaired differentiation). Blue triangle: FLT3 ligands. Brown circles: extracellular membrane. Green arrows: FLT3-mediated leukemogenic activity in the absence of FLT3 inhibitors; red arrows: impairment of FLT3-mediated leukemogenic activity in the presence of FLT3 inhibitors. Abbreviations: FLT3, FMS-like tyrosine kinase; TKD, tyrosine kinase domain; ED, extramembrane domain; TMD, transmembrane domain; JMD, juxtamembrane domain.
2. Combination Regimens Including Gilteritinib in R/R and De Novo Acute Myeloid Leukemia
2.1. Gilteritinib Plus Azacitidine in FLT3-Mutated AML
2.1. Gilteritinib Plus Azacitidine in FMS-Related Tyrosine Kinase 3-Mutated Acute Myeloid Leukemia
Wang et al. [25] proposed a randomized phase 3 trial aimed to assess the efficacy and safety of gilteritinib plus azacitidine vs. azacitidine in newly diagnosed FLT3-mutated AML considered not eligible for intensive chemotherapy. Patients were randomized (2:1) to be treated with gilteritinib (120 mg/day orally) and azacitidine at standard dosage or azacitidine alone on a 28-day cycle. In all, 123 patients were enrolled, 74 included in the gilteritinib–azacitidine arm (median age, 78 years) and 49 in the azacitidine arm (median age 76 years); among them, 47.3% and 32.7% had an ECOG performance status (PS) of 2 in the two arms, respectively.
Authors found no significant difference in OS between the two arms; the median OS was 9.82 months and 8.87 months, respectively (HR 0.916; 95% CI, 0.529–1.585; p = 0.753). The median EFS was 0.03 months in both treatment arms; the CRc rate was significantly higher in the gilteritinib–azacitidine arm than in the azacitidine arm (58.1% and 26.5%, respectively; p < 0.001). Furthermore, authors observed a numeric improvement in OS with gilteritinib–azacitidine in some patient subgroups, but statistical significance was not reached. In the subgroup of patients stratified as having an ECOG PS of 0 to 1, the median OS was 13.17 months and 11.89 months, respectively (HR, 0.811; 95% CI, 0.409–1.608; p = 0.549); among patients with an FLT3–ITD allelic ratio of 0.5 or higher, the median OS was 10.68 months and 4.34 months, respectively (HR, 0.580; 95% CI, 0.285–1.182; p = 0.134). AE rates were similar between the arms. AEs of any grade occurred in 100% of patients in the gilteritinib–azacitidine arm and 95.7% of those in the azacitidine arm. The rate of grade 3 or higher AEs was 95.9% and 89.4%, respectively [26]. According to these data, this combination approach did not improve survival outcomes in patients, with newly diagnosed FLT3-mutated AML unfit for intensive treatment. Therefore, the trial was closed based on the protocol-specified boundary for futility and recommendations from the independent data monitoring committee.
2.2. Gilteritinib Plus Venetoclax in R/R AML
2.2. Gilteritinib Plus Venetoclax in R/R Acute Myeloid Leukemia
Venetoclax has been approved as a standard treatment in combination with low-dose cytarabine or hypomethylating agents for newly diagnosed AML ineligible for intensive chemotherapy [27,28]. Single-agent venetoclax showed limited activity in R/R AML [29]; however, in vitro reports demonstrated synergistic activity between venetoclax and FLT3 inhibitors in preclinical models [30,31].
In an American, multicenter study, 61 patients with R/R AML, including 56 with FLT3-mutated disease, were enrolled to receive a combination regimen based on venetoclax and gilteritinib; 15 patients were enrolled in the dose-escalation phase and 46 were enrolled in the dose-expansion phase. The trial provided 400 mg of venetoclax once daily and gilteritinib at 80 mg or 120 mg once daily during dose escalation, with the recommended phase II dose being venetoclax at 400 mg and gilteritinib at 120 mg. Among the 56 patients with FLT3-mutated disease treated at any dose, after a median follow-up of 17.5 months, the modified composite CR (consisting of complete response, complete response with incomplete blood count recovery, complete response with incomplete platelet recovery, and morphologic leukemia-free state) rate was 75% (the CR rate was 18%). The median time to response and median remission duration was 0.9 months and 4.9 months, respectively, with a median OS of 10.0 months. Modified composite CR was observed in 14 (67%, CR in 29%) of 21 patients with no prior FLT3 TKI exposure and in 28 (80%, CR in 11%) of 35 patients with prior TKI exposure. The median OS was 10.6 months and 9.6 months, respectively. Grade 3 or 4 AEs occurred in 97% of patients, mostly characterized by cytopenias (80%). AEs led to venetoclax and gilteritinib interruptions in 51% and 48% of patients and to discontinuation of treatment in 15% and 13%, respectively. Serious AEs occurred in 75% of patients, most commonly febrile neutropenia (44%) and pneumonia (13%) [32]. This combination approach produced a highly modified composite CR rate in patients with FLT3-mutated R/R AML; however, dose interruptions for cytopenias were very common, and this regimen showed a high toxicity profile.
The addiction of gilteritinib to azacitidine and venetoclax in FLT3-mutated AML was another fascinating triplet combination. In the phase I/II trial recently reported by Short et al., the ORR was 100% (27/27), with a 92% CR in newly diagnosed patients, a median OS that had not yet been attained, and an OS of 85% at 1 year. In R/R patients, the ORR was 70% (14/20), with a CR rate of 20% (4/20) and a median OS of 5.8 months. With a median OS of 10.5 months, outcomes were better in patients who had not previously received gilteritinib or venetoclax [33].
2.3. Gilteritinib Plus Chemotherapy in Patients with Newly Diagnosed AML
2.3. Gilteritinib Plus Chemotherapy in Patients with Newly Diagnosed Acute Myeloid Leukemia
Recently, encouraging data on the association between gilteritinib and induction and consolidation chemotherapy were presented at the 10th Annual Meeting of the Society of Hematologic Oncology. Patients enrolled in this phase 1 trial (NCT02236013) were required to be at least 18 years of age with newly diagnosed AML and have an ECOG performance status of 2 or less; the presence of an FLT3 mutation at baseline was not required. Dose escalation of gilteritinib was assessed in part 1 of the study to identify the MTD. Induction regimen provided 3 days of idarubicin with 7 days of cytarabine and 14 days of gilteritinib at doses of 20 mg, 40 mg, 80 mg, 120 mg, or 200 mg, given on days 4 through 17 for up to 2 cycles. The consolidation approach included high-dose cytarabine plus the same dose of gilteritinib given daily for the first 14 days of each cycle for up to 3 cycles. Finally, patients received maintenance treatment based on gilteritinib daily for 28 days for up to 26 cycles. The dose expansion study (part 2) provided gilteritinib at 120 mg a day, with induction, consolidation, and maintenance following the same treatment pattern as dose expansion trial. In part 3 of the study, the gilteritinib dosing schedule during induction was modified to begin with the completion of chemotherapy, running from days 8 through 21, and the other receiving 3 days of daunorubicin and 7 days of cytarabine. Consolidation and maintenance followed the same treatment pattern as parts 1 and 2. In part 4 of the study, gilteritinib was given up to 56 consecutive days during consolidation. A total of 79 patients were enrolled; among them, 56.4% of patients harbored FLT3 mutations, 42.3% had FLT3–ITD mutations, and 41% had FLT3wt disease. At the end of treatment, the composite CR in patients with FLT3 mutation was 90.9%, with 70.6% of patients achieving a CR. The 26-week, 1-year, and 2-year OS rates were 92.4%, 82.1%, and 69.2%, respectively, in this subgroup. Additional data showed that while censoring for HSCT, the median disease-free survival (DFS) for patients with FLT3 mutations (n = 40) was 460 days (95% CI, 150–970), while the FLT3-negative population (n = 22) experienced a median DFS of 288 days (95% CI, 23–971). The MTD of gilteritinib was established to be 120 mg per day, and dose-limiting toxicities occurred in 15 of 78 (19.2%) patients given gilteritinib. AEs led to the discontinuation of gilteritinib in 24.4% of patients. Grade ≥ 3 treatment-emergent AEs were reported in 93.6% of patients [34]. According to these results, an effective antileukemic response was observed in terms of CR and OS, particularly in the FLT3-mutated subgroup in newly diagnosed AML who received gilteritinib in combination with intensive chemotherapy. These data support further trials to confirm the validity of this approach and to compare this regimen with the already approved treatment based on the combination of midostaurin with intensive chemotherapy in FLT3-mutated patients. Table 1 summarizes the trials including gilteritinib for the treatment of de novo AML. Table 2 summarizes the ongoing and recruiting studies including gilteritinib in combination with chemotherapy or other small molecules in R/R and de novo AML.
Table 1.
Trials including gilteritinib for the treatment of de novo acute myeloid leukemia.
| Number of Patients | Median Age | Response | Mrdian Duration of Response | Median EFS 1/DFS 2 |
Survival | Number of Reference | |
|---|---|---|---|---|---|---|---|
| 5-Aazacitidine + Gilteritinib | 74 | 78 years (range 59–90) | CRc 3 58.1% | 8.57 months | Median EFS 1 4.53 months | Median OS 5 9.82 months | [26] |
| 5-Aazacitidine + Venetoclax + Gilteritinib |
21 | 68 years (range, 18–82) | ORR 4 100% | Not reported | Not reported | 1-year OS 5 rate 85% | [33] |
| Standard chemotherapy (induction and consolidation) + Gilteritinb | 44 | 50 years (range 23–77) | CRc 3 90.9% | Not reported | Median DFS 2 460 days | 1-year OS 5 rate 82.1% 2-year OS 5 rate 69.2% |
[34] |
1 EFS = event-free survival. 2 DFS = disease-free survival. 3 CRc = composite complete remission. 4 ORR = overall response rate. 5 OS = overall survival.
Table 2.
Ongoing and recruiting studies including gilteritinib.
| Number of the Study | Protocol Regimen | Eligible Patients |
|---|---|---|
| NCT04027309 | gilteritinib vs. midostaurin in combination with induction and consolidation therapy followed by one-year maintenance | newly diagnosed acute myeloid leukemia or myelodysplastic syndromes with excess blasts-2 with FLT3 mutations |
| NCT04140487 | azacitidine, venetoclax, and gilteritinib | relapsed/refractory FLT3-mutated acute myeloid leukemia, chronic myelomonocytic leukemia, or high-risk myelodysplastic syndrome/myeloproliferative neoplasm |
| NCT04240002 | gilteritinib combined with chemotherapy | children, adolescents, and young adults with FLT3–ITD-positive relapsed/refractory acute myeloid leukemia |
| NCT05546580 | iadademstat and gilteritinib | relapsed/refractory acute myeloid leukemia with FLT3–ITD mutation |
| NCT05520567 | gilteritinib, venetoclax, and azacitidine | newly diagnosed with acute myeloid leukemia with FLT3 mutations |
| NCT05028751 | lanraplenib (lanra) in combination with gilteritinib | FLT3-mutated relapsed/refractory acute myeloid leukemia |
| NCT05010122 | astx727, venetoclax, and gilteritinib | newly diagnosed, relapsed/refractory FLT3-mutated acute myeloid leukemia or high-risk myelodysplastic syndrome |
| NCT04293562 | standard chemotherapy vs. therapy with cpx-351 and/or gilteritinib | newly diagnosed acute myeloid leukemia with or without FLT3 mutations |
| NCT05010772 | decitabine alone or in combination with venetoclax, gilteritinib, enasidenib, or ivosidenib as maintenance therapy | acute myeloid leukemia in remission |
3. Gilteritinib for Extramedullary AML Relapse
The FLT3–ITD gene mutation has been described to promote leukemic cell infiltration into visceral organs while inhibiting homing to the bone marrow by downregulation of CXCR4 signaling [43]. Several studies have demonstrated that miRNAs may promote hematopoiesis and hematological diseases [42,43]. FLT3-mediated signaling controls the expression of several miRNAs, with both downregulation (miR-451 and miR-144) and upregulation (miR-155, miR-10a, and miR-10b) mechanisms. The expression of these small molecules seems to favor extramedullary blasts infiltration, although underlying mechanisms remain to be demonstrated [44]. Several case reports have described the efficacy of gilteritinib in patients with FLT3 mutant extramedullary relapse before or after transplant. Perrone et al. first demonstrated the potential biological effect of gilteritinib within the central nervous system (CNS) [45]. Moreover, Vignal et al. reported the presence of gilteritinib in cerebrospinal fluid at therapeutic doses [46]. In another case [47], a patient experienced a right supraclavicular mass with simultaneously occurring AML blasts relapsed after ASCT in the bone marrow. Both extramedullary and medullary blasts presented FLT3–ITD mutation. Therapy with 120 mg/day of gilteritinib was started, determining a medullary and extramedullary CR and allowing the patient to proceed to a second HSCT [47]. Furthermore, gilteritinib seems to have efficacy also in infrequent localization of AML. Kim et al. [48] described a case of an FLT3–ITD-mutated patient who presented an AML relapse involving the temporal iris, ciliary body, and choroid by a leukemic infiltrative mass. The patient started treatment with oral gilteritinib, obtaining rapid regression of the tumor, with a significant improvement in visual acuity [48]. The mechanisms underlying the documented activity of gilteritinib in extramedullary AML are still unknown; however, this small molecule appears to hold efficacy in this setting and should be taken into consideration, especially in heavily pretreated patients.
4. Antifungal Prophylaxis in Patients Treated with Gilteritinib
Due to the fact that gilteritinib mainly undergoes CYP3A4-dependent metabolism, the manufacturer advises against using gilteritinib concurrently with drugs that strongly induce or inhibit CYP3A4 and instead suggests to select alternative treatments [10]. In the phase I/II CHRYSALIS trial, which examined possible drug–drug interactions between gilteritinib and moderate and strong CYP3A4 inhibitors (such as fluconazole, voriconazole, and posaconazole), gilteritinib exposure was found to be less than two times higher when an azole was also administered. The incidence of AEs did not vary between patients who received a moderate or strong CYP3A4 inhibitor and those who did not; hence, this increase was not deemed to be clinically relevant [17]. The effects of weak CYP3A4 inhibitors (such as itraconazole) and strong CYP3A4 inhibitors (such as fluconazole) on the pharmacokinetics of gilteritinib were assessed in an open-label drug–drug interaction research study. The findings showed that fluconazole was associated with a smaller increase in systemic exposure to gilteritinib (1.43-fold) compared with itraconazole (2.3-fold), which was linked with a significant increase in systemic exposure to gilteritinib [10]. The larger phase III ADMIRAL trial, however, forbade the use of posaconazole, itraconazole, and voriconazole, leaving unaddressed the issue on how to combine these drugs [8]. Aleissa et al. assessed the prevalence of AEs associated with gilteritinib in 47 patients who received gilteritinib either with or without antifungal triazoles. In the gilteritinib–triazole group, AEs related to gilteritinib were comparable to those in the gilteritinib group without triazole (75% vs. 55.5%, p = 0.23). The severity of AEs, dose reductions or discontinuations from gilteritinib (15% vs. 14.8%), and 90-day mortality (35% vs. 11.1%) were also comparable between the two groups [49]. However, how interactions between azoles and gilteritinib impact toxicities is not yet fully defined. Therefore, the European Hematology Association guideline on antifungal prophylaxis in patients with AML treated with novel-targeted therapies recommended triazole antifungal prophylaxis for patients who are heavily pretreated with gilteritinib [50].
5. Development of Resistances to Gilteritinib
Around 30% of patients who relapse after achieving a remission to type 1 FLT3 inhibitors (midostaurin, gilteritinib, and crenolanib) carry mutations in the RAS pathway, making it the most prevalent mutation-derived mechanism of resistance to type 1 inhibitors. These mutations may appear as new mutations following therapy or as clonal proliferation with rising variant allele frequency (VAF) over the course of therapy [51]. Poorer outcomes in both primary and secondary relapse scenarios are linked to higher VAFs in RAS/MAPK mutations. RAS pathway mutations are less common with type 2 FLT3 inhibitors (quizartinib) than with type 1 inhibition, occurring in just 6% of patients relapsing after type 2 inhibitors. RAS-mutated clones can spread in patients using quizartinib, even though FLT3–TKD mutations are the most common route of resistance to type 2 inhibitors [52]. It was hypothesized that the preservation of FLT3 mutant clones can also depend on the bone marrow microenvironment (BMME). Indeed, soluble cytokines and growth factors together with cell–cell contact between leukemic cells and stromal cells within BMME can act as a mediator for the preservation of leukemic clones [53]. BMME adaptation and changes have been described alongside therapy. Patients relapsing after intensive chemotherapy courses were found to have considerably greater FLT3 ligand levels, inducing AKT, ERK, and other proapoptotic proteins’ downregulation through FLT3 ligand-FLT3wt binding. Despite FLT3–ITD inhibition, FLT3wt-mediated activation of these pathways promotes leukemic cell survival [54].
In the ADMIRAL study, 40 patients acquired new mutations during treatment. Among them, in 18 patients, the RAS/MAPK pathway was affected, while FLT3 was involved in 6 cases (5 patients presented the F691L mutation); 3 had WT1 (1 had the F691L mutation), 1 had IDH1, and 1 had GATA2. Thirteen patients (32.5%) had no new mutations. During relapse, FLT3 F691L gatekeeper mutations and mutations in the RAS/MAPK pathway genes were mutually exclusive [55]. RAS/MAPK and FLT3 F691L mutations were acquired by nontransplanted patients during relapse; however, the latter did not correlate with refractoriness. Uncertainty exists regarding the relationship between the dosage of gilteritinib and the prevalence of emergent FLT3 F691L gatekeeper mutations at relapse. In the ADMIRAL study, patients who received gilteritinib at 120 mg/day had a comparable incidence of FLT3 F691L, as seen in relapsed patients who received gilteritinib from 20 to 200 mg/day, but none of the patients receiving >200 mg/day acquired this kind of mutation at relapse. However, compared with other patients, those receiving 120 mg/day had improved OS [56]. Another study demonstrated a relationship between gilteritinib dose and occurrence of resistance in 22 FLT3-mutated patients analyzed at relapse by next-generation sequencing and single-cell analysis, reporting a more likely onset of RAS or FLT3 F691L mutations in those treated with doses below 200 mg [57].
Recently, it was reported that FF-10101, a selective and irreversible FLT3 inhibitor, significantly inhibited FLT3–ITD and -TKD mutations, including F691L and D835, both in vitro and in vivo [58,59]. Fifty-two patients with R/R AML were enrolled in a phase I dose escalation study to test the inhibitor. In pretreated patients (median number of prior therapies, n = 3), continuous treatment with FF-10101 at a dose of 10–225 mg 4 times per day or 50–100 mg twice daily led to a composite CR rate of 13% and a partial response rate of 8%, including those with activating FLT3–TKD mutations resistant to gilteritinib and other FLT3 TKIs. Well-tolerated doses of 50–75 mg twice daily resulted in long-lasting FLT3 suppression. The trial is still ongoing but not recruiting patients [60].
Sitravatinib is a multikinase inhibitor under evaluation in ongoing clinical trials of several solid tumors. In a recent study, the antitumor activity of sitravatinib against FLT3–ITD and clinically relevant drug resistance in FLT3 mutant AML were explored. The FLT3–ITD-F691L mutation caused resistance to gilteritinib and all other FLT3 inhibitors, both in vitro and in vivo, whereas sitravatinib showed a potent inhibitory impact. With stronger and more consistent suppression of p-ERK and p-AKT than gilteritinib, sitravatinib maintained excellent efficacy against FLT3 mutation in the presence of cytokines. Additionally, sitravatinib was more effective against patient blasts carrying FLT3–ITD in vitro and in the PDX model than gilteritinib [61].