Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jason Zhu and Version 3 by Fanny Huang.
While chemotherapies remain the mainstay treatments for many cancers, the advent of new molecular techniques has opened doors for more targeted modalities towards cancer cells. Although immune checkpoint inhibitors (ICIs) have demonstrated therapeutic efficacy in treating cancer, adverse side effects related to excessive inflammation are often reported. There is a lack of clinically relevant animal models to probe the human immune response towards ICI-based interventions. Humanized mouse models have emerged as valuable tools for pre-clinical research to evaluate the efficacy and safety of immunotherapy.
- humanized mouse model
- cancer
- immunotherapy
1. Introduction
Most cancers are classified into immune-hot and immune-cold groups based on their immune infiltrations, with immunotherapy showing more significant effects on the supposedly immune-hot groups as seen in the case of melanoma [1]. Despite the safety of some drugs in certain cancers, they may cause adverse reactions or unsuccessful treatment in others. For example, lung airways or gut-associated lymphoid tissues have different immune environments from those of the liver or pancreas; this could be a reason for the failure of targeted therapies [2]. A well-developed humanized mouse model can overcome these limitations and serve as a preclinical platform for testing the safety and efficacy of new and existing drugs, along with characterising adverse events and identifying the best sequence of immunotherapy agents for therapy. Using a humanized mouse model allows researchers to test the antitumour cytotoxic responses of immune cells and assess the safety of a drug in the presence of both tumours and immune systems. Performing a pharmacokinetic (PK) and/or bioavailability study in humanized mice over time, and with different dosages, can help clinical scientists evaluate the stability of a given drug in patients and safely identify an evidence-based dosing regimen for that drug [3]. As different patients may respond differently to the same drug, using peripheral blood mononuclear cells (PBMCs) from patients that are to be treated could be used to predict the reactions of these patients to the drug, thus presenting a path to personalised medicine.
Essentially, a humanized mouse model is one that has human immune cells engrafted into immunodeficient mice. There are three commonly known methods of generating these humanized models (Figure 1), which include inoculation with PBMCs, inoculation with CD34+ hematopoietic stem cells (HSCs) alone, or simultaneously engraftment with human foetal liver and thymic tissue (BLT) into the sub-renal capsules of immunocompromised mice. Engraftment with purified human PBMCs via intraperitoneal injection into homozygous severe combined immunodeficient model (SCID) mice was first performed in 1988 [4]. Over time, immunodeficient models were improved (Table 1), and advanced NSG (Il2rgnull) mice, which show a high chimerism rate, the development of human erythropoiesis and thrombopoiesis, and the formation of haemato-lymphoid compartments, have become the bases for the development of today’s humanized mice models [5][6].
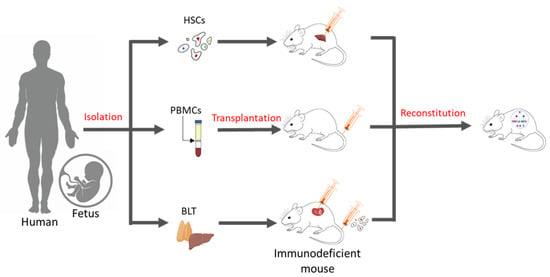
Figure 1. Methods of generating humanized mice. Humanized mouse models may be generated in three ways: (a) By the intra-hepatic injection of adult or foetal HSCs in immunodeficient mice, (b) by administering human PBMCs via tail-vein injections, and (c) through the simultaneous transplantation of human foetal liver and thymus tissue under kidney capsules and the intravenous injection of human CD34+ HSCs in immunodeficient mice.
Table 1. Most commonly used immunodeficient mice models. Much progress has been made in the development of immunodeficient models for the engraftment of human immune cells. While earlier models also supported the engraftment of PBMCs and HSCs, the reconstitution and chimerism in these models were low, possibly due to the effects of residual murine cells. This table summarises the most commonly used immunodeficient models for different applications along with their distinct characteristics.
| Models | Mutations | Advantages | Disadvantages | Refs. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SCID | Protein kinase DNA-activated catalytic peptide gene ( | Prkdc | ) | Lack humoral and cell-mediated immunity Absence of B and T cells |
Leakiness due to age Reduced lifespan Presence of murine NK cells Complementation of activity |
[4][7] | ||||
| NOD/SCID | Signal regulatory protein alpha ( | Sirpa | ) | High affinity to human CD47Tolerance of host macrophages to the human cellsImmunological multi-dysfunction, including defective NK cell activitySupport higher levels of human engraftments | Residual NK cell activity, and some other innate compounds of the immune system, fail to develop mature monocytes, which eventually become thymic lymphomas with age |
[8][9][10][11][12][13] | ||||
| BRG Rag1/2 |
Recombination-activating gene 1 ( | Rag1 | ) or 2 ( | Rag2 | ) | No leakiness Absence of functional B and T cellsLonger lifespans |
Limited lymphoid reconstitution Residual NK cell activity |
[14] | ||
| NOG NSG |
Interleukin-2 receptor gamma chain ( | IL2r | γ) null phenotype and Prkdc mutation (NOD/SCID-IL2Rγ | −/− | ) | Absence of functional receptors for cytokines like IL-2 and IL-7 Could hamper the development of host NK cells Highest rate of human cell engraftments Longer lifespans |
Weak human myeloid reconstitution Lack of human thymus tissue Radiation sensitivity |
[5][6][12][15] | ||
| NRG | RAG1/2 null mutation and Interleukin-2 receptor gamma chain ( | IL2r | γ) null (NOD/RAG1/2 | −/− | - IL2Rγ | −/− | ) | Improved myeloid engraftmentCan tolerate chemotherapy at higher doses | Higher radiation dose for preconditioning Lack of human thymus tissue Possible background activity |
[16] |
| FRG | Fah/Rag2/IL-2rγ | Triple knockout Expand human hepatocytes robustly Gene and cell therapy |
Remnant mouse hepatocytes Background activity |
[17][18] |
The possibility of PBMCs and tumour tissues originating from the same patients proved to be a key advantage of the Hu-PBMC model, but resulted in only a partial reconstitution of the human immune system in the peripheral blood of the mice, with most cells being CD3+ T cells that eventually interacted with major histocompatibility complex (MHC) molecules in mice and led to graft-versus-host disease (GvHD) [4]. This incomplete reconstitution could be improved via the engraftment of HSCs characterised by the expression of the cell surface marker CD34, which is obtained from umbilical cord blood, peripheral blood, or the foetal liver. These models showed higher leukocyte reconstitution and greater engraftment efficacy than other sources and provided insights into the organisation of the human stem-cell hierarchy and bases for functional assays, which had been difficult to achieve with the earlier models [19]. However, their use was impeded as mice from the NOD/SCID strain appeared to develop a limited number of B and T cells, in a phenomenon called leakiness, during aging [20]. These mice also had higher levels of host murine NK cells, with significantly low human NK cell counts—possibly to compensate for the lack of lymphocytes—which eventually hampered the engraftment of human cells [19]. Human T cells undergo the process of human leukocyte antigen (HLA)-restriction and T cell selection in the thymus, which enables the T-cell receptor (TCR) to recognise and bind to certain MHC molecules that present foreign antigens and initiate a T-cell immune response. NSG mice, however, express the H2 complex, not HLA molecules, on their thymic epithelial cells, and thus the T cells in the HSC model lack HLA-restriction. This can be overcome with the advancement of the BLT mouse model, allowing for T cell maturation in an autologous human thymus, overcoming the murine H2 restriction, and promoting human T cells that are capable of HLA-restricted antigen-specific reactions [21][22]. Despite these ideal conditions, the BLT model is technically challenging to develop and may lead to immunologic rejection due to the existence of mismatched HLA between immune cells and tumour tissues. The discussed models also lack human NK cells, and the need for foetal tissue has raised several ethical issues.
A broad spectrum of specific humanized oncology models has been generated, including those for human nasopharyngeal cancer (NPC) [23], hepatocellular carcinoma (HCC) [24], gastric carcinoma [25], colon carcinoma [25], prostate cancer [26], bladder cancer [27], head and neck squamous cell carcinoma (HNSCC) [28], lymphoma [29], and melanoma [30][31]. These can be either cell-line-derived (CDX) or PDX models. Researchers lab has been at the forefront of establishing cancer-specific humanized mouse models in Southeast Asia and has demonstrated the importance of specificity in these models, pointing out that the immune system can have different effects on the TMEs of different cancers. For example, the growth of the tumour in the NPC humanized model in the presence of the human immune system is significantly inhibited, whereas in the HCC model, the size and weight of tumours are increased as compared to what one would find in the immunodeficient mice [23][24].
Humanized mouse models allow detailed studies of the TME and tumour cells’ interactions with the human immune system, providing deeper insight into the mechanisms of tumour progression, metastasis, and development of immunotherapies. They have been used in studies on different immunotherapies including antibody-based ICI, adoptive cellular therapy (ACT, which encapsulates methods like CAR-T and CAR-NK cell therapy), cytokine manipulation, cancer vaccination, and the development of oncolytic viruses (Figure 2).
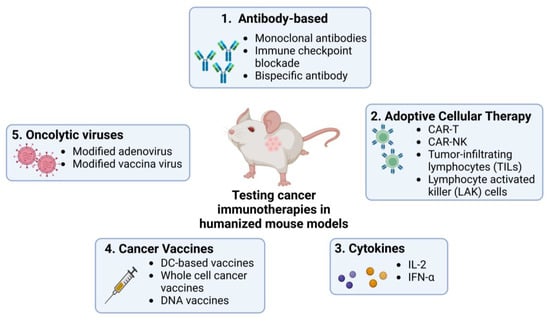
Figure 2. Use of humanized mouse models to test current immunotherapies in cancer research. Many immunotherapies, including ICIs, ACT, cytokine-based therapies, and therapies using cancer vaccines and oncolytic viruses, are currently being studied using humanized mouse models. These models provide platforms for testing the efficacy and safety of the commonly known therapies mentioned in the figure in human-like systems. Created by BioRender.com.
2. Humanized Mouse Models for Testing ICIs and Antibody-Based Drugs
Increasingly, evidence of dysregulated levels of immune checkpoints in tumour-infiltrated T cells has been noted, thus posing these cells as attractive targets for immunotherapies. These checkpoints include cytotoxic T-lymphocyte antigen 4 (CTLA-4), programmed cell death protein 1 (PD-1), lymphocyte activation gene 3 (LAG-3), and T-cell immunoglobulin and mucin domain 3 (TIM-3), as seen in Figure 3 [32].
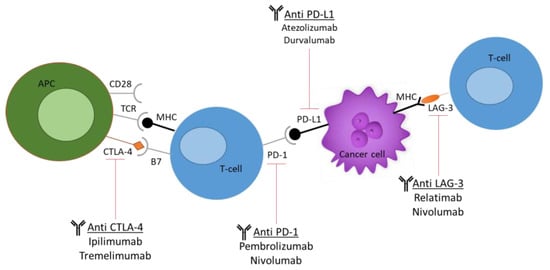
Figure 3. Current immune checkpoint inhibitors (ICIs) used in cancer research. The most commonly used ICIs, including anti-PD-1, anti-PD-L1, anti-CTLA-4, and anti-LAG-3. The figure demonstrates the molecular and cellular interactions between the T cells and cancer cells and the immune response activation upon antibody administration.
ICIs form a part of monoclonal antibody (mAb) targeted therapy. Humanized mouse models play an important role in the preclinical study and testing of ICIs and their interactions with tumour-infiltrating lymphocytes (TILs), thus helping in predicting the efficacy of these drugs as seen in Table 2.
The HCC and NPC humanized mouse models established in researchers lab were able to successfully validate existing anticancer and combinatorial therapies [23][24][33]. These models successfully showed that cancer cells educate the immune cells and condition the TME to suppress the immune system and aid in tumorigenesis. The HCC model also replicated the immunotoxicity caused by ipilimumab (anti-CTLA-4 antibody) in patients [24], while an exploration of the NPC model highlighted similarities between clinical data from patients and the results from this model [23][34]. Both models indicated the presence of an exhaustive phenotype [34] with the presence of TILs and the upregulation of inhibitory receptors, thus establishing a human-specific platform for drug testing and a path to using these models for discovering new therapeutic strategies and drug combinations.
Further, a PDX humanized model for non-small cell lung cancer (NSCLC) has indicated the hCD8 T-cell-mediated efficacy of pembrolizumab (anti-PD-1 antibody) and shown the significant growth inhibitory effect of pembrolizumab on these tumours [35]. Similarly, a sarcoma PDX humanized mouse model showed consistently elevated levels of hCD8+ T cells and their subsets, implying the effect of these cells in antitumour activity, with reductions in tumour size [36]. This model also recapitulated the sarcoma TME as seen in clinical findings and predicted the NK-related factors that affect the survival of patients [36]. These findings show the importance of these models in verifying cellular and molecular targets for drug development and hint towards the mechanisms associated with their actions.
Table 2. Safety and efficacy profiles of a few ICI-based therapies. A brief description of some cancers that were modelled in humanized mice and used for testing the safety and efficacy of human-specific ICI-based drugs.
| Mouse model | Target | Safety | Efficacy | Model Description | Refs. | |||
|---|---|---|---|---|---|---|---|---|
| Colon | CD137 and PD-1 | √ | √ | CDX-model with intraperitoneal injection of PBMCs in Rag2 | −/− | IL2Rγnull strain | [25] | |
| Gastric | CD137 and PD-1 | √ | √ | PDX-model with intraperitoneal injection of PBMCs in Rag2 | −/− | IL2Rγnull strain | [25] | |
| HCC | 1 | PD-1 and CTLA-4 | √ | √ | PDX-model with intrahepatic injection of human CD34+ HSCs in NSG mice | [24][33] | ||
| NPC | 2 | PD-1 and CTLA-4 | √ | √ | PDX-model with intrahepatic injection of human CD34+ HSCs in NSG mice | [23] | ||
| Lymphoma | PD-1 and CTLA-4 | √ | √ | CDX-model with subcutaneous engraftment of human PBMCs in NOG mice | [37][38] | |||
| Sarcoma | PD-1 | √ | √ | PDX-model with intravenous injection of human CD34+ CB cells in NSG mice | [36] | |||
| NSCLC | 3 | PD-1 | √ | √ | CDX and PDX-models with intravenous injection of human CD34+ HPSCs in NSG mice | [35][39] | ||
| PD-L1 | √ | √ | CDX and PDX-models with intravenous injection of either PBMCs or human CD34+ HSCs in NSG mice | [40] | ||||
| Bladder | PD-1 | √ | √ | PDX-model with intravenous injection of human CD34+ HPSCs in NSG mice | [35] | |||
| TNBC | 4 | PD-1 | √ | √ | CDX and PDX-models with intravenous injection of human CD34+ HPSCs in NSG mice | [35] | ||
| Lung adenocarcinoma | PD-1 | √ | √ | CDX-model with intravenous injection of CD34+ human HSCs in NOG and NOG-FcγR | −/− | mice | [41] | |
| PD-L1 | √ | √ | CDX-model with intravenous injection of cord-blood derived CD34+ human HSCs in NSG mice | [42] | ||||
| HNSCC | 5 | PD-1 | √ | √ | CDX-model with intravenous injection of CD34+ human HSCs in NOG and NOG-FcγR | −/− | mice | [41] |
| Ovarian carcinoma | PD-L1 | √ | √ | CDX-model with intravenous injection of fetal liver-derived CD34+ human HSCs in NOG mice | [42] |
1 HCC Hepatocellular Carcinoma. 2 NPC Nasopharyngeal Carcinoma. 3 NSCLC Non-Small Cell Lung Cancer. 4 TNBC Triple-Negative Breast Cancer. 5 HNSCC Head and Neck Squamous Cell Carcinoma.
3. Safety and Efficacy Profiling of ACT in Humanized Mouse Models
With the hype surrounding personalised medicine, ACT has gained much importance with respect to cancer immunotherapy. It has indeed shown promising results, especially with respect to metastatic melanoma [43] and B-cell malignancies [44]. This method involves identifying and isolating immune cells or lymphocytes from a patient, expanding them ex vivo, and then infusing them back into the cancer patient, targeting the tumour cells, and resulting in antitumour activity. However, treatment-related toxicities have limited their widespread implementation in therapeutic regimes, with experimental studies being irreproducible in patients. With the use of humanized mouse models, these challenges are being addressed with enhanced treatment efficacy prediction.
3.1. CAR-T Cells
CARs are constructed by integrating the single-chain fragment variable (scFv) domain of an antibody with the TCR constant domain, presenting the TCRs with high affinity and specificity for target antigens. This allows the activation of modified T cells in an MHC-independent manner, thus bypassing the challenge posed by tumour escape [45]. CAR-T therapy, especially when involving anti CD19-CAR-T cells, has shown impressive results in clinical trials involving B-cell malignancies [46][47] and multiple myeloma [48]. However, only approximately half the patients in these trials were shown to respond, while some acquired resistance and suffered major toxicities including CRS, neurologic toxicity, and B-cell aplasia, thereby limiting the potential of this therapy [44]. A humanized mouse model could be used not only to design innovative CAR constructs and develop novel CAR-T cell therapies but also to evaluate their safety and efficacy against disease.
Considering the severe toxicities of CRS in ACT, murine models mimicking CRS in patients have been developed to generate a better understanding of CRS pathophysiology and thereby improve the safety, and reduce the toxicities, of CAR-T therapies. A study using SCID-beige mice to model CRS reported serum cytokine levels that were similar to those reported in clinical studies and established the important role of macrophage-derived IL-6, IL-1, and nitric oxide (NO) in the pathophysiology of CRS [49]. Genetically engineered CAR-T cells that constitutively produce IL-1 receptor antagonist were shown to protect from CRS-mortality along with maintaining the antitumour efficacy in NSG mice; this finding is useful when attempting to identify a new target for CRS and also to design a novel CAR construct [49]. This genetic engineering technology can be boosted by silencing unwanted or faulty genes to provide optimal therapeutic effects. This technique was recently employed to silence PD-1 expression in CAR-T cells and has showed improved polyfunctionality of CAR-T in vitro and in PDX models [50]. The functionality of silencing PD-1 and gene editing techniques can be further investigated in humanized mouse models. As such, gene-edited CAR-T cells with disrupted PD-1 signalling were shown to have enhanced activity in humanized CDX-derived glioblastoma mouse models [51]. The utilised method prompts the development of allogeneic CAR-T cells to provide off-the-shelf therapy rather than relying on autologous CAR-T cells.
Following these studies, a B-cell acute lymphoblastic leukaemia (ALL) humanized mouse model was used to generate CD-19-targeted CAR-T cells from human autologous mature T cells and successfully recapitulated the response seen in patients, demonstrating its efficacy and providing an insight into a potential mechanism behind CRS, with granulocyte macrophage colony-stimulating factor (GM-CSF), a pro-inflammatory cytokine, playing a key role in CRS following CAR-T therapy [52]. Such models can be highly valuable in evaluating the efficacy of various CAR-T cells in the treatment of other malignancies, improving safety and reducing resistance by illustrating the underlying mechanisms of newly established treatments.
3.2. CAR-NK Cells
The potential of CAR-NK cells in allogenic cell therapy has emerged in recent years due to the desired properties of these cells such as lower incidence of GvHD, accessibility, and short lifespan in vivo, making them more manageable than CAR-T cell therapy as part of an alternative approach to cancer treatment [53].
Researchers lab employed third-generation CAR NK92 cell line, which has been shown to target programmed cell death ligand-1 (PD-L1) and display improved cytotoxicity and apoptosis in various human cell lines that express PD-L1, in the NPC humanized mouse model in order to optimise the use of these cells in treating solid cancers and potentially induce their antitumour activity [54]. Indeed, the injection of HSC-derived primary CAR-NK (CAR-pNK) cells in humanized mice showed the stimulation of the antigen processing and presenting pathways, indicating the activation of the host immune system despite the presence of exhaustive TILs. Furthermore, treatment regimens involving CAR-pNK cells and nivolumab (anti-PD-1 antibody) were designed, and these showed synergistic antitumour responses in the humanized mice, thus demonstrating that CAR-NK therapy can be further pursued for the treatment of NPC and potentially other solid tumours [54].
Another study using genetically engineered cord-blood-derived CAR-NK cells expressing IL-15 and an inducible caspase-9 suicide gene showed improved antitumour toxicity in a CDX lymphoma humanized mouse model. Interestingly, the activation of the suicide gene was able to eliminate the CAR-NK cells in vivo efficiently and rapidly in cases of associated toxicities, and the expression of IL-15 helped in the conservation and proliferation of the stem cell memory T-cell phenotype with an improved antitumour function. These anti-CD19 CAR-NK cells are currently being evaluated in clinical trials, thus establishing the humanized mouse model as a bridge to translational medicine [55].
4. Improving Cytokine-Based Immunotherapy Using Humanized Mouse Models
Cytokines evoke biological functions in the immune system and can enhance the proliferation and survival of T cells and NK cells upon activation. Cytokines like IL-2 have pleiotropic functions, such as proliferating CD8+ T cells and enhancing the cytotoxic activity, while also stimulating regulatory T cells (Tregs), which are associated with the suppression of antitumour response [56]. Cytokine-mediated immunotherapy using an antitumor cytokine called interferon-alpha 2 (IFN-α2) was the first immunotherapy agent approved by the Food and Drug Administration (FDA) in 1986, and it is understood that the use of humanized mice was extremely limited at the time. IL-2 is one of the few cytokines that has been approved in immunotherapy by the FDA, first for the treatment of metastatic renal cell carcinoma in 1992 and then for the treatment of metastatic melanoma in 1998 [57]. Since these studies were conducted prior to the discovery of fully humanized mice, they showed the survival of human and murine T cells in vitro and in wild-type mice and were translated into clinical testing, where they resulted in dose-related toxicities like fever, chills, malaise, and mild hepatic dysfunction while having no effect on tumour regression [58]. Several other studies followed suit, having similar conclusions [59][60][61].
To improve the efficacy of these cytokine-mediated therapies using humanized mouse models, others have explored the potential of a humanized mouse model of ovarian cancer to study the TME and to test the efficacy of IL-12 targeted immunotherapy. While these studies showed no significant decreases in tumour progression upon treatment with liposome-mediated cytokines, they showed elevations in the levels of IFN-γ in the experimental mice, indicating that T lymphocytes and possibly NK cells are both viable and responsive to IL-12 stimulation and thereby providing evidence of the potential of this treatment [62].
5. Immune Response of Cancer Vaccines in Humanized Mouse Models
Following the coronavirus disease 2019 (COVID-19) pandemic, the importance of vaccines has been highlighted. Scientists have been trying to integrate the theory of vaccinations into the treatment and prevention of cancers. Similar to COVID-19 vaccines, cancer vaccines aim to train the body’s immune system to identify and destroy potentially harmful tumour cells, and can be divided into virus-, peptide-, nucleic-acid-, or cell-based vaccines depending on preparation method. Prophylactic vaccines targeting human papilloma virus and hepatitis B virus (HBV) have gained FDA-approval and are widely implemented in clinics to prevent malignancies associated with viral pathogens [63], while therapeutic vaccines are still underway.
Tumour progression is often accompanied by the presence of somatic mutations. When these mutations occur in protein-coding genes, they introduce non-synonymous polymorphisms which may give rise to novel tumour neoantigens [64]. Tumour antigens recognised by T lymphocytes are essential for ensuring cancer vaccine efficacy and to activate the immune system. These mainly include tumour-associated antigens or tumour-specific antigens. Humanized mouse models can be utilised to screen for such neoantigens and further design optimal targeted vaccines. Multiple dendritic cell (DC) vaccines have been generated and studied in humanized mice. One study compared three DC vaccine formulations using a melanoma antigen recognised by T-cells 1 (MART-1) as an antigen, allowing for the selection of the combination that was most robust and showed an enhanced immune response in a 3-day period [65]. Additionally, a novel mAb (HuMAb006-11) against HBV, which is a commonly known cause of HCC, was developed in a human liver chimeric mouse model. This mAb protected humanized mice from an active HBV infection, inhibited viral entry, and assisted in clearing the virions from circulation, thus establishing its prophylactic effects [66]. These studies highlighted the use of humanized mouse models in proposing novel candidates for future clinical translation and selection of the most optimum therapeutic regimen of cancer vaccines.
6. Targeted Tumour Lysis by Oncolytic Viruses (OVs) Using Humanized Mice
OV immunotherapy enlists organisms that identify, infect, and lyse tumour cells in the TME, aiming to halt or decrease tumour progression by activating DCs with damage-associated molecular patterns (DAMPs) and tumour antigens while sparing surrounding healthy cells. These organisms could be either genetically engineered or naturally occurring viruses. Their use in clinics is gaining importance, with the first FDA approval for oncolytic vaccines being granted in 2015 [67]. They can be used as single agents or in combination with other therapies and have contributed to tumour cell death and increased the overall efficacy of immunotherapies [67]. However, the clinical manifestations of this therapy are highly variable, demonstrating a need for the humanized mouse model. While the injection of OVs into solid tumours in mouse models has been tested and has showed promising results [68][69], studying the interaction between the virus-colonised tumours and human immune system is highly important in order to understand the optimal mode of action of this therapy, which has emphasised the importance of the humanized mouse model.
A humanized glioma-PDX mouse model has demonstrated not only the efficacy of using OVs, such as CRAd-S-pK7, for malignant gliomas, but also a novel intranasal delivery system using CXCR4-enhanced neural stem cells that leads to the tumour-specific delivery of OVs and extends the survival time of test animals [70].
The interactions of the oncolytic vaccinia virus with tumours were analysed using humanized mice bearing A549 (human lung carcinoma) cell lines. This model demonstrated successful selection, infection, and replication of the virus in the tumours in vivo post administration of the GLV-2b372 oncolytic vaccinia virus strain in the humanized tumour-bearing mice, and further indicated targeted tumour cell lysis. Additional studies have investigated the combination of OVs with ICIs in syngeneic murine models and have demonstrated robust immune-mediated antitumour activity [71], with improved results to be established using humanized mouse models.
7. Combination Therapy with ICIs
Despite the advantages of immunotherapy, there are some sub-groups that do not respond favourably to a single cancer immunotherapy agent. The treatment of solid cancers has proven to be a challenge with their varied populations, poor tissue penetration, and heterogenous populations, leading to drug resistance mechanisms being developed. For such cases, combining different immunotherapy drugs or administering therapeutic antibodies in conjunction with chemotherapeutics and other modes of treatment has been shown to be more effective in enhancing antitumour efficacy [72]. Clinical trials testing the effects of combination therapy involving two ICIs, anti-CTLA-4 and anti-PD-1, have shown promising results in cancers like melanoma and have already been approved by the FDA for clinical use [73], and other trials using combinations of relatlimab (anti-LAG-3 antibody) and nivolumab are already underway [74]. However, these combinatorial therapies often exhibit adverse immune-related side effects due to a lack of proper understanding of the synergy between the incorporated compounds and their possible overlap in other cellular mechanisms. This has hampered the development of new combination therapies and provides an avenue for the humanized mouse model to be of use.
The NPC humanized mouse model was used to test the efficacy of a combination treatment of nivolumab and ipilimumab and showed results consistent with those seen in donor patients [23]. The association of the Epstein–Barr Virus (EBV) with NPC and other cancers like Burkitt’s Lymphoma, Hodgkin Lymphoma, and NK/T cell lymphoma led to a study using HSC-humanized NRG mice injected intravenously with EBV and targeted with adoptively transferred T cells specific for EBV alone and in combination with nivolumab [75]. The upregulation of checkpoint inhibitors in the study model indicated a strong affinity for combination therapy, which was associated with significantly increased survival and reduced tumour burden in tumour-bearing mice, establishing the importance of combination therapy.
Using the HCC immune system–humanized mouse model, researchers lab showed that combination therapy in the form of dual and triple therapy could be used to validate anti-HCC effects and side effects in humanized mice. Triple therapy using pembrolizumab, bevacizumab (anti-vascular endothelial growth factor (VEGF)), and the Stat inhibitor C188-9 resulted in an improved combinatorial effect, with the lowest level of proliferation and evidence of reduced angiogenesis, thus significantly increasing the efficacy of the anti-HCC response compared with the results of traditional therapy [32]. Another study using HCC-PDX humanized mouse model demonstrated the anti-HCC effect of anti-GPC3 CAR-T cell therapy [76], providing more evidence on the importance of these models for the testing of novel combinatorial therapeutic strategies.
More recent studies have demonstrated the immunosuppressive and exhaustive phenotypes of ovarian cancers and demonstrated the efficacy of using dual blockade with anti-LAG-3 and anti-PD-1 in murine models with a significantly higher number of T cells in the TME, enhanced CD8+ TIL functions, and a reduced subset of Treg cells [77]. One of the more promising results for treating ovarian cancer was discovered by combining a PD-1/CTLA-4 dual blockade with the adoptive transfer of autologous TILs in humanized PDX-ovarian cancer models.
A prior study using a humanized mouse model engrafted with triple-negative breast cancer (TNBC) cell lines showed the relevance of combination therapy for the treatment of TNBC with nivolumab and OKI-179 (histone deacetylase inhibitor) [78]. This treatment significantly inhibited tumour growth and allowed for the dose modification of nivolumab after a certain period. Hence, it not only provided new therapeutic approaches to TNBC but also validated the efficacy of humanized mouse models in the evaluation of immunotherapy and of combination therapies. Combination therapy has been applied in the humanized oncology model by showing the use of temozolomide (a chemotherapy drug) and ibudliast (a macrophage migration inhibitory factor inhibitor) in extending the survival of mice in a glioblastoma-PDX model [79].
The extensive range of studies using these models for testing and evaluating the different components of immunotherapy proves the impact of these models in advancing the field of personalised medicine and oncology (Table 3). The use of human PBMCs and human HSCs helps not only to create a platform to study and understand the effect of these immunotherapies, but also to discover new targets that could be utilised to develop anticancer agents and provide personalised immune-oncology models.
Table 3. The use of humanized mouse models in different immunotherapies, Summarising their applications and safety and efficacy profiles along with indications on how they can be improved to overcome their current limitations and provide an enhanced platform for drug testing and development.
| Immunotherapy | Humanized Mouse Model Application | Safety & Efficacy | Further Modifications | Limitations |
|---|---|---|---|---|
| Antibody-based | Help in discovering new targets for ICIs | Improved compared to wild type mice | Immune-transgenic models | Undefined resistance mechanisms |
| Adoptive Cell Therapy | Hints towards possible mechanisms underlying CRS | Higher safety and efficacy standards | Improved NK cell model for cytotoxic activity | CRS and neurotoxicity, limited effect on solid tumours |
| Cytokine | Gaining popularity to prevent tumour growth | Potential to improve | Fully humanized models with improved cytokine transgenic models | Lack of specificity |
| Cancer vaccines | Prophylactic and therapeutic vaccines are being established using this model | Dose escalation studies provide improved safety and efficacy | Dual humanized mouse models for specific viral cancers | Scarcity of suitable neoantigens |
| Oncolytic viruses | Discovering novel delivery systems and targeting tumour lysis | Improved safety and efficacy when translated to clinic | Dual humanized mouse models | Need for improved delivery strategies, tumour heterogeneity |
References
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 11, 197–218.
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 1, 11.
- Flora, A. Evidence-Based Selection of the Starting Dose in First-in-Human Clinical Trials Using Humanized Mouse Models. The Jackson Laboratory. Available online: https://www.jax.org/news-and-insights/jax-blog/2022/april/evidence-based-selection-starting-dose-in-human-clinical-trials (accessed on 15 February 2023).
- Mosier, D.E.; Gulizia, R.J.; Baird, S.M.; Wilson, D.B. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature 1988, 331, 256–259.
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005, 171, 6477–6489.
- Ishikawa, F.; Yasukawa, M.; Lyons, B.; Yoshida, S.; Miyamoto, T.; Yoshimoto, G.; Watanabe, T.; Akashi, K.; Shultz, L.D.; Harada, M. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor chain(null) mice. Blood 2005, 101, 1565–1573.
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A severe combined immunodeficiency mutation in the mouse. Nature 1983, 301, 527–530.
- Makino, S.; Kunimoto, K.; Muraoka, Y.; Mizushima, Y.; Katagiri, K.; Tochino, Y. Breeding of a non-obese, diabetic strain of mice. Exp. Anim. 1980, 21, 1–13.
- Mian, S.A.; Anjos-Afonso, F.; Bonnet, D. Advances in Human Immune System Mouse Models for Studying Human Hematopoiesis and Cancer Immunotherapy. Front. Immunol. 2021, 11, 619236.
- Mullen, Y. Development of the Nonobese Diabetic Mouse and Contribution of Animal Models for Understanding Type 1 Diabetes. Pancreas 2017, 41, 455–466.
- Takenaka, K.; Prasolava, T.K.; Wang, J.C.; Mortin-Toth, S.M.; Khalouei, S.; Gan, O.I.; Dick, J.E.; Danska, J.S. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat. Immunol. 2007, 1, 1313–1323.
- Shultz, L.D.; Schweitzer, P.A.; Christianson, S.W.; Gott, B.; Schweitzer, I.B.; Tennent, B.; McKenna, S.; Mobraaten, L.; Rajan, T.V.; Greiner, D.L.; et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. 1995, 151, 180–191.
- Serreze, D.V.; Gaedeke, J.W.; Leiter, E.H. Hematopoietic stem-cell defects underlying abnormal macrophage development and maturation in NOD/Lt mice: Defective regulation of cytokine receptors and protein kinase C. Proc. Natl. Acad. Sci. USA 1993, 91, 9625–9629.
- Ménoret, S.; Fontanière, S.; Jantz, D.; Tesson, L.; Thinard, R.; Rémy, S.; Usal, C.; Ouisse, L.H.; Fraichard, A.; Anegon, I. Generation of Rag1-knockout immunodeficient rats and mice using engineered meganucleases. FASEB J. 2013, 21, 703–711.
- Lee, J.Y.; Han, A.R.; Lee, D.R. T Lymphocyte Development and Activation in Humanized Mouse Model. Dev. Reprod. 2019, 21, 79–92.
- Shultz, L.D.; Banuelos, S.; Lyons, B.; Samuels, R.; Burzenski, L.; Gott, B.; Lang, P.; Leif, J.; Appel, M.; Rossini, A.; et al. NOD/LtSz-Rag1nullPfpnull mice: A new model system with increased levels of human peripheral leukocyte and hematopoietic stem-cell engraftment. Transplantation 2003, 71, 1036–1042.
- Azuma, H.; Paulk, N.; Ranade, A.; Dorrell, C.; Al-Dhalimy, M.; Ellis, E.; Strom, S.; Kay, M.A.; Finegold, M.; Grompe, M. Robust expansion of human hepatocytes in Fah−/−/Rag2−/−/Il2rg−/− mice. Nat. Biotechnol. 2007, 21, 903–910.
- Chen, J.; Li, Y.; Lai, F.; Wang, Y.; Sutter, K.; Dittmer, U.; Ye, J.; Zai, W.; Liu, M.; Shen, F.; et al. Functional Comparison of Interferon-α Subtypes Reveals Potent Hepatitis B Virus Suppression by a Concerted Action of Interferon-α and Interferon-γ Signaling. Hepatology 2021, 71, 486–502.
- Lapidot, T.; Fajerman, Y.; Kollet, O. Immune-deficient SCID and NOD/SCID mice models as functional assays for studying normal and malignant human hematopoiesis. J. Mol. Med. 1997, 71, 664–673.
- Bosma, G.C.; Fried, M.; Custer, R.P.; Carroll, A.; Gibson, D.M.; Bosma, M.J. Evidence of functional lymphocytes in some (leaky) scid mice. J. Exp. Med. 1988, 161, 1016–1033.
- Yin, L.; Wang, X.J.; Chen, D.X.; Liu, X.N.; Wang, X.J. Humanized mouse model: A review on preclinical applications for cancer immunotherapy. Am. J. Cancer Res. 2020, 11, 4568–4584.
- Cogels, M.M.; Rouas, R.; Ghanem, G.E.; Martinive, P.; Awada, A.; Van Gestel, D.; Krayem, M. Humanized Mice as a Valuable Pre-Clinical Model for Cancer Immunotherapy Research. Front. Oncol. 2021, 11, 784947.
- Liu, W.N.; Fong, S.Y.; Tan, W.W.S.; Tan, S.Y.; Liu, M.; Cheng, J.Y.; Lim, S.; Suteja, L.; Huang, E.K.; Chan, J.K.Y.; et al. Establishment and Characterization of Humanized Mouse NPC-PDX Model for Testing Immunotherapy. Cancers 2020, 11, 1025.
- Zhao, Y.; Shuen, T.W.H.; Toh, T.B.; Chan, X.Y.; Liu, M.; Tan, S.Y.; Fan, Y.; Yang, H.; Lyer, S.G.; Bonney, G.K.; et al. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut 2018, 61, 1845–1854.
- Sanmamed, M.F.; Rodriguez, I.; Schalper, K.A.; Oñate, C.; Azpilikueta, A.; Rodriguez-Ruiz, M.E.; Morales-Kastresana, A.; Labiano, S.; Pérez-Gracia, J.L.; Martín-Algarra, S.; et al. Nivolumab and Urelumab Enhance Antitumor Activity of Human T Lymphocytes Engrafted in Rag2−/−IL2Rγnull Immunodeficient Mice. Cancer Res. 2015, 71, 3466–3478.
- Roth, M.D.; Harui, A. Human tumor infiltrating lymphocytes cooperatively regulate prostate tumor growth in a humanized mouse model. J. Immunother. Cancer 2015, 1, 12.
- Pan, C.X.; Shi, W.; Ma, A.H.; Zhang, H.; Lara, P.; Keck, J.G.; Palucka, K.; Airhart, S.D.; White, R.D. Humanized mice (humice) carrying patient-derived xenograft (PDX) as a platform to develop immunotherapy in bladder cancer (BCa). Clin. Oncol. 2017, 31 (Suppl. S6), 381.
- Morton, J.J.; Bird, G.; Keysar, S.B.; Astling, D.P.; Lyons, T.R.; Anderson, R.T.; Glogowska, M.J.; Estes, P.; Eagles, J.R.; Le, P.N.; et al. XactMice: Humanizing mouse bone marrow enables microenvironment reconstitution in a patient-derived xenograft model of head and neck cancer. Oncogene 2016, 31, 290–300.
- Ito, A.; Ishida, T.; Yano, H.; Inagaki, A.; Suzuki, S.; Sato, F.; Takino, H.; Mori, F.; Ri, M.; Kusumoto, S.; et al. Defucosylated anti-CCR4 monoclonal antibody exercises potent ADCC-mediated antitumor effect in the novel tumor-bearing humanized NOD/Shi-scid, IL-2Rgamma(null) mouse model. Cancer Immunol. Immunother. 2009, 51, 1195–1206.
- Tsoneva, D.; Minev, B.; Frentzen, A.; Zhang, Q.; Wege, A.K.; Szalay, A.A. Humanized Mice with Subcutaneous Human Solid Tumors for Immune Response Analysis of Vaccinia Virus-Mediated Oncolysis. Mol. Ther. Oncolytics 2017, 1, 41–61.
- Schupp, J.; Christians, A.; Zimmer, N.; Gleue, L.; Jonuleit, H.; Helm, M.; Tuettenberg, A. In-Depth Immune-Oncology Studies of the Tumor Microenvironment in a Humanized Melanoma Mouse Model. Int. J. Mol. Sci. 2021, 21, 1011.
- Franzin, R.; Netti, G.S.; Spadaccino, F.; Porta, C.; Gesualdo, L.; Stallone, G.; Castellano, G.; Ranieri, E. The Use of Immune Checkpoint Inhibitors in Oncology and the Occurrence of AKI: Where Do We Stand? Front. Immunol. 2020, 11, 574271.
- Zhao, Y.; Wang, J.; Liu, W.N.; Fong, S.Y.; Shuen, T.W.H.; Liu, M.; Harden, S.; Tan, S.Y.; Cheng, J.Y.; Tan, W.W.S.; et al. Analysis and Validation of Human Targets and Treatments Using a Hepatocellular Carcinoma-Immune Humanized Mouse Model. Hepatology 2021, 71, 1395–1410.
- He, Q.F.; Xu, Y.; Li, J.; Huang, Z.M.; Li, X.H.; Wang, X. CD8+ T-cell exhaustion in cancer: Mechanisms and new area for cancer immunotherapy. Brief Funct. Genom. 2019, 11, 99–106.
- Wang, M.; Yao, L.C.; Cheng, M.; Cai, D.; Martinek, J.; Pan, C.X.; Shi, W.; Ma, A.H.; De Vere White, R.W.; Airhart, S.; et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J. 2018, 31, 1537–1549.
- Choi, B.; Lee, J.S.; Kim, S.J.; Hong, D.; Park, J.B.; Lee, K.Y. Anti-tumor effects of anti-PD-1 antibody, pembrolizumab, in humanized NSG PDX mice xenografted with dedifferentiated liposarcoma. Cancer Lett. 2020, 471, 56–69.
- Donnou, S.; Galand, C.; Touitou, V.; Sautès-Fridman, C.; Fabry, Z.; Fisson, S. Murine models of B-cell lymphomas: Promising tools for designing cancer therapies. Adv. Hematol. 2012, 2011, 701704.
- Ma, S.D.; Xu, X.; Jones, R.; Delecluse, H.J.; Zumwalde, N.A.; Sharma, A.; Gumperz, J.E.; Kenney, S.C. PD-1/CTLA-4 Blockade Inhibits Epstein-Barr Virus-Induced Lymphoma Growth in a Cord Blood Humanized-Mouse Model. PLoS Pathog. 2016, 12, e1005642.
- Qiao, T.; Xiong, Y.; Feng, Y.; Guo, W.; Zhou, Y.; Zhao, J.; Jiang, T.; Shi, C.; Han, Y. Inhibition of LDH-A by Oxamate Enhances the Efficacy of Anti-PD-1 Treatment in an NSCLC Humanized Mouse Model. Front. Oncol. 2021, 11, 632364.
- Lin, S.; Huang, G.; Cheng, L.; Li, Z.; Xiao, Y.; Deng, Q.; Jiang, Y.; Li, B.; Lin, S.; Wang, S.; et al. Establishment of peripheral blood mononuclear cell-derived humanized lung cancer mouse models for studying efficacy of PD-L1/PD-1 targeted immunotherapy. MAbs 2018, 11, 1301–1311.
- Katano, I.; Hanazawa, A.; Otsuka, I.; Yamaguchi, T.; Mochizuki, M.; Kawai, K.; Ito, R.; Goto, M.; Kagawa, T.; Takahashi, T. Development of a novel humanized mouse model for improved evaluation of in vivo anti-cancer effects of anti-PD-1 antibody. Sci. Rep. 2021, 11, 21087.
- Li, Y.; Carpenito, C.; Wang, G.; Surguladze, D.; Forest, A.; Malabunga, M.; Murphy, M.; Zhang, Y.; Sonyi, A.; Chin, D.; et al. Discovery and preclinical characterization of the antagonist anti-PD-L1 monoclonal antibody LY3300054. J. Immunother. Cancer 2018, 1, 31, Erratum in J. Immunother. Cancer 2018, 1, 45.
- Rosenberg, S.A.; Dudley, M.E. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr. Opin. Immunol. 2009, 21, 233–240.
- Vanegas, Y.M.; Mohty, R.; Gadd, M.E.; Luo, Y.; Aljurf, M.; Qin, H.; Kharfan-Dabaja, M.A. CAR-T cell Therapies for B-cell Lymphoid Malignancies: Identifying Targets Beyond CD19. Hematol. Oncol. Stem Cell Ther. 2022, 11, 81–93.
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 91, 720–724.
- Marofi, F.; Achmad, H.; Bokov, D.; Abdelbasset, W.K.; Alsadoon, Z.; Chupradit, S.; Suksatan, W.; Shariatzadeh, S.; Hasanpoor, Z.; Yazdanifar, M.; et al. Hurdles to breakthrough in CAR T cell therapy of solid tumors. Stem Cell Res. Ther. 2022, 11, 140.
- Havard, R.; Stephens, D.M. Anti-CD19 chimeric antigen receptor T cell therapies: Harnessing the power of the immune system to fight diffuse large b cell lymphoma. Curr. Hematol. Malig. Rep. 2018, 11, 534–542.
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 121, 1688–1700.
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 21, 731–738.
- Diorio, C.; Murray, R.; Naniong, M.; Barrera, L.; Camblin, A.; Chukinas, J.; Coholan, L.; Edwards, A.; Fuller, T.; Gonzales, C.; et al. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood 2022, 141, 619–629.
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfo, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 1, 304.
- Jin, C.H.; Xia, J.; Rafiq, S.; Huang, X.; Hu, Z.; Zhou, X.; Brentjens, R.J.; Yang, Y.G. Modeling anti-CD19 CAR T cell therapy in humanized mice with human immunity and autologous leukemia. EBioMedicine 2019, 31, 173–181.
- Li, H.; Song, W.; Li, Z.; Zhang, M. Preclinical and clinical studies of CAR-NK-cell therapies for malignancies. Front. Immunol. 2022, 11, 992232.
- Liu, W.N.; So, W.Y.; Harden, S.L.; Fong, S.Y.; Wong, M.X.Y.; Tan, W.W.S.; Tan, S.Y.; Ong, J.K.L.; Rajarethinam, R.; Liu, M.; et al. Successful targeting of PD-1/PD-L1 with chimeric antigen receptor-natural killer cells and nivolumab in a humanized mouse cancer model. Sci. Adv. 2022, 8, eadd1187.
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 31, 520–531.
- Chulpanova, D.S.; Kitaeva, K.V.; Green, A.R.; Rizvanov, A.A.; Solovyeva, V.V. Molecular Aspects and Future Perspectives of Cytokine-Based Anti-cancer Immunotherapy. Front. Cell Dev. Biol. 2020, 1, 402.
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in cancer immunotherapy. Oncoimmunology 2016, 5, e1163462.
- Lotze, M.T.; Frana, L.W.; Sharrow, S.O.; Robb, R.J.; Rosenberg, S.A. In vivo administration of purified human interleukin 2. I. Half-life and immunologic effects of the Jurkat cell line-derived interleukin 2. J. Immunol. 1985, 131, 157–166.
- Lotze, M.T.; Matory, Y.L.; Ettinghausen, S.E.; Rayner, A.A.; Sharrow, S.O.; Seipp, C.A.; Custer, M.C.; Rosenberg, S.A. In vivo administration of purified human interleukin 2. II. Half life, immunologic effects, and expansion of peripheral lymphoid cells in vivo with recombinant IL 2. J. Immunol. 1985, 131, 2865–2875.
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 191, 5451–5458.
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Chang, A.E.; Avis, F.P.; Leitman, S.; Linehan, W.M.; Robertson, C.N.; Lee, R.E.; Rubin, J.T.; et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N. Engl. J. Med. 1987, 311, 889–897.
- Bankert, R.B.; Balu-Iyer, S.V.; Odunsi, K.; Shultz, L.D.; Kelleher RJJr Barnas, J.L.; Simpson-Abelson, M.; Parsons, R.; Yokota, S.J. Humanized mouse model of ovarian cancer recapitulates patient solid tumor progression, ascites formation, and metastasis. PLoS ONE 2011, 6, e24420.
- Stanley, M. Tumour virus vaccines: Hepatitis B virus and human papillomavirus. Philos. Trans. R Soc. Lond. B Biol. Sci. 2017, 371, 20160268.
- Roudko, V.; Greenbaum, B.; Bhardwaj, N. Computational Prediction and Validation of Tumor-Associated Neoantigens. Front. Immunol. 2020, 11, 27.
- Spranger, S.; Frankenberger, B.; Schendel, D.J. NOD/scid IL-2Rg(null) mice: A preclinical model system to evaluate human dendritic cell-based vaccine strategies in vivo. J. Transl. Med. 2012, 11, 30.
- Jhajharia, S.; Lai, F.; Low, H.B.; Purushotorman, K.; Shunmuganathan, B.D.; Chan, C.E.Z.; Hammond, R.; Netter, H.J.; Chen, Q.; Lim, S.G.; et al. Defining the specificity and function of a human neutralizing antibody for Hepatitis B virus. NPJ Vaccines 2022, 1, 121.
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 1, 841–849.
- Yu, Y.A.; Shabahang, S.; Timiryasova, T.M.; Zhang, Q.; Beltz, R.; Gentschev, I.; Goebel, W.; Szalay, A.A. Visualization of tumors and metastases in live animals with bacteria and vaccinia virus encoding light-emitting proteins. Nat. Biotechnol. 2004, 21, 313–320.
- Zhang, Q.; Yu, Y.A.; Wang, E.; Chen, N.; Danner, R.L.; Munson, P.J.; Marincola, F.M.; Szalay, A.A. Eradication of solid human breast tumors in nude mice with an intravenously injected light-emitting oncolytic vaccinia virus. Cancer Res. 2007, 61, 10038–10046.
- Dey, M.; Yu, D.; Kanojia, D.; Li, G.; Sukhanova, M.; Spencer, D.A.; Pituch, K.C.; Zhang, L.; Han, Y.; Ahmed, A.U.; et al. Intranasal Oncolytic Virotherapy with CXCR4-Enhanced Stem Cells Extends Survival in Mouse Model of Glioma. Stem Cell Rep. 2016, 1, 471–482.
- Minev, B.; Kohrt, H.; Kilinc, M.; Chen, N.; Feng, A.; Pessian, M.; Geissinger, U.; Haefner, E.; Tsoneva, D.; Bozhilov, K.; et al. Combination immunotherapy with oncolytic vaccinia virus and checkpoint inhibitor following local tumor irradiation. J. Immunother. Cancer 2014, 2 (Suppl. S3), P112.
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 1, 86.
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 361, 122–133, Erratum in N. Engl. J. Med. 2018, 371, 2185.
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. RELATIVITY-047 Investigators. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 381, 24–34.
- Sinha, D.; Srihari, S.; Beckett, K.; Le Texier, L.; Solomon, M.; Panikkar, A.; Ambalathingal, G.R.; Lekieffre, L.; Crooks, P.; Rehan, S.; et al. ‘Off-the-shelf’ allogeneic antigen-specific adoptive T-cell therapy for the treatment of multiple EBV-associated malignancies. J. Immunother. Cancer 2021, 9, e001608.
- Jiang, Z.; Jiang, X.; Chen, S.; Lai, Y.; Wei, X.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; Liang, Q.; et al. Anti-GPC3-CAR T Cells Suppress the Growth of Tumor Cells in Patient-Derived Xenografts of Hepatocellular Carcinoma. Front. Immunol. 2017, 1, 690.
- Huang, R.Y.; Francois, A.; McGray, A.R.; Miliotto, A.; Odunsi, K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology 2016, 6, e1249561.
- Capasso, A.; Lang, J.; Pitts, T.M.; Jordan, K.R.; Lieu, C.H.; Davis, S.L.; Diamond, J.R.; Kopetz, S.; Barbee, J.; Peterson, J.; et al. Characterization of immune responses to anti-PD-1 mono and combination immunotherapy in hematopoietic humanized mice implanted with tumor xenografts. J. Immunother. Cancer 2019, 1, 37.
- Ha, W.; Sevim-Nalkiran, H.; Zaman, A.M.; Matsuda, K.; Khasraw, M.; Nowak, A.K.; Chung, L.; Baxter, R.C.; McDonald, K.L. Ibudilast sensitizes glioblastoma to temozolomide by targeting Macrophage Migration Inhibitory Factor (MIF). Sci. Rep. 2019, 1, 2905.
More