Carbon anode refers to a broad family of essentially pure carbon, whose members can be tailored to vary widely in their strength, density, conductivity, pore structure, and crystalline development. These attributes contribute to their widespread applicability. Specific characteristics are imparted to the finished product by controlling the selection of precursor materials (including cokes, polymers and fibers) and the method of processing. In general, carbon anode electrodes are characterized by low cost production, high surface area, a wide working potential window in many media, high electrocatalytic activities for different redox-active chemical and biochemical systems, and chemical inertness.Moreover, their surface chemistry enables the functionalization of these carbon platforms via strong covalent or noncovalent methods with surface modifiers, which improves their electrochemical performance. Recent achievements of carbon anode materials and their structural design for better performances of aluminium production, lithium-ion secondary batteries, lithium cobalt oxide batteries, nano-tube production, substitution of amorphous electrode materials, photoanodes production, solar cells, fuel cells, supercapacitors, sensors and pumps, neurochemical monitors, etc., are finding enormous applications in industrial, commercial and social sectors.
- carbon electrode materials
- aluminium production
- electrode kinetics
- energy storage technologies
- neurochemical monitoring.
1. Introduction
At the start of things, that is more than 15 Gy ago, all the matter and energy that we can observe was concentraded in a volume element about the size of a small coin (~100 mm3). Later, within resultant stars, at temperatures of about 1015 K, hydrogen atoms were stripped of their nuclei and fused to form helium nuclei. As stars cooled, collision of helium nuclei led to beryllium, of fleeting stability but of sufficient stability to allow a further collision with a helium nucleus, to give us carbon. Also, a continuing collision of carbon with a helium nucleus gave us oxygen; and so our story has started.
Some 5 Gy ago, from of the cosmic dust out there, an event occurred leading to the formation of the solar system, with the sun, planets, and moons. Only one planet (earth) was of the correct size and at an optimal distance from the sun to create and maintain oceans and an atmosphere. The atmosphere was first made up by volcanic activity, of carbon dioxide and water vapour, two greenhouse gases. Eventually, nitrogen was added to the atmosphere. The oceans were created and carbon dioxide was established within the carbon cycle with formation of carbonates. These greenhouse gases kept the atmospheric and oceanic temperatures compatible with chemical reactions leading to molecules evolution, i.e., the creation of life. The oceanic plant forms took out the carbon dioxide from the atmosphere and replace it with oxygen (photosynthesis). These early plants became entrapped within the rocks and their remains are now identified as kerogens, and petroleum deposits and methane reservoirs. As the continents of earth moved over its surface the great rainforests were established, which provided the organic material that, after being chemically degraded and fossilised, is now recognizable as coal. Nature in the process of maturation of kerogen and coal, had, as the end product, graphitic material, which is the basis of all carbon forms, with the exception of crystalline diamond. About 15,000 years ago, petroleum and coal appeared making life very easy for modern man. The waste products of the petroleum industry (the distillation of the barrel), and the waste products of the coal carbonization industry (the manufacture of metallurgical coke) as coal-tar and coal-tar pitch, are used to create the carbon artefacts (matrix and binder) of the carbon electrode industry[1] [1].
The development of the several segments of the carbon industry can be seen to be quite closely related. They did not develop independently of each other, really. The years after WW-II saw extremely rapid advances in mass production of consumer goods and in mass transportation. A starting point for our story could be with Henry Ford and his motor cars (Figure 1).
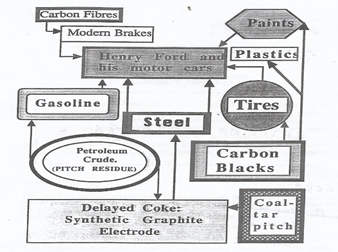
Figure 1. A schematic illustrating the interdependence of carbon industries.
The automobile had, at least, two basic requirements; it needed metal for its production and petrol (gasoline) for its mobility. Iron metal production soared, coking plants were built to feed the blast furnaces, and coal-tar and coal-tar pitch were indeed plentiful (a glut). Petroleum companies were growing in number and size and gasoline availability soared. However, not all of the barrel could be converted to gasoline or jet fuels, or fuel oils. The remnants of the barrel, asphaltenes, petroleum pitch residue, had no commercial value and presented a disposable problem. Land fill was out of the question and use as a fuel was impractical (unlike coal-tar pitch). The delayed coker was developed to take care of growth in amounts of disposable pitch and the words ‘delayed coke’ were heard more frequently. Landfill and use as fuel were more practical with a solid.
During the first half of past century, it had become obvious that the route to aluminium production was via the Hall-Héroult cell, i.e., the electrochemical reduction of alumina, by carbon, in a molten bath of cryolite. Developments of the carbon anode had pointed the way to the use of a coke bonded with coal-tar pitch. At the same time as the aluminium industry was expanding, the petroleum and steel making industries were providing the necessary ingredientes of anode manufacture. Of course, refinements in the quality of residues going to the delayed coker were necessary (to make regular and needle coke, as distinct from shot coke), and more stringent specifications were applied to coal-tar pitch quality. However, one situation has not changed over all of these years, which is that the aluminium industry has to cope with the problems of quality control of its essential supplies, considered by the suppliers as waste materials. There is an additional complication in that petroleum and coal resources are changing with exploitation and hence continuous quality control of coke and pitch for the anode is a necessity [2][3][4][5][2–5].
In terms of the history of the carbon industries, carbon blacks impinge into our story. Although dominantly associated with printing inks, carbon blacks are an essential ingredient of the automobile tyre. Aircraft use braking systems of carbon composites made up of carbon fiber matrices bonded with carbon from coal-tar pitch. Also, the steel industry makes its steels in the furnace heated using the grafite electrode, made from premium quality delayed coke (needle coke) and coal-tar pitch. In the present century, sophisticated carbon electrodes have been manufactured for many applications, namely in the area of electrochemical energy devices[6][7][8] [6–8].
Light metal meetings and many other meetings involving several industries using carbon electrodes continue to show that there are many factors that need to be considered and improved to obtain efficient anode electrodes. Structure and purity of delayed cokes, permeability, the role of catalytic impurities, wettability by the pitch, the physical properties of the pitch (e.g., the relevance of glass transition temperatures), and the role of QI material in pitch all continue to dominate the discussions. Restricting to the aluminium electrowinning, we really do not understand very well what happens within the green anode when we pyrolyse and bake it, namely we do not know exactly how to moderate optimum relationships between coke particle (shape and size), butt particle, and mixing extent with coke particles, and the shape and size of the binder coke bridges[9] [9]. During pyrolysis and baking, I suspect that we have little true idea of the interactions that are occurring between the pitch and, later, the mesophase derived from pitch with the particulate components of the green paste. Summarizing, there is still a great future for the light metals meetings and related meetings on carbon materials.
At this point, it seems that it is time to stress that not all days are black in the carbon world. Looking back at the carbon highligts, we clearly find some areas deserving attention. It is the case of the aluminium production in large alumina refineries, using carbon anodes of high quality, which depends on the characteristics of coke filler, coal tar pitch binder, and anode scrap, among others[5] [5]. However, then we can see the development of synthetic diamonds by the GE high-pressure catalytic process initiated in 1941 and leading to the first commercially successful synthesis on december 1954[10][11] [10,11]. Much later the diamond and diamondlike films appeared, using low temperature and low pressure, truly defiant of all the laws of thermodynamics and phase diagrams[12][13] [12,13]. The carbon fibers, emerging first from PAN, and later from pitch, are other excellent carbon materials whose development led to the carbon fiber reinforced plastic (CFRP) and other composite products, which have several uses in aerospace and non-aerospace structures, as well as in non-structural applications (thermal insulation, electrodes for batteries and supercapacitors, hydrogen gas storage, etc.)[14][15][16][17] [14–17]. One of the most exciting events of all has been the explanation for the formation of anisotropic, graphitizable carbons via the intermediate phase of mesophase, that nematic, aromatic, discotic liquid crystal system[18] [18]. However, probably the most intriguing discovery has been the fullerene systems and the nanotubes, including curved crystals, inorganic fullerenes and nanorods, hybrids of carbon nanotubes and graphene, carbon anions and spheroidal carbon particles, and other related nanosctructures that are capturing the imagination of physicists, chemists, materials scientists, and nanotechnologists alike[19][20] [19–22]. These new discoveries and developments had an impact that extends well beyond the confines of academic research and worked the beginning of a new era in carbon science and technology[21][22][23][24] [23–26]. In this century, the progress is still slow, but applications begin to appear, and future prospects are enormous. Moreover, the field of carbon electrochemistry has experienced a robust development over the last decades with the emergence of the multidimensional carbon materials cited above[24][25] [26,27].
In general, carbon-based electrodes are characterized by low cost production, high surface areas, a wide working potential window in many media, high electrocatalytic activities for different redox-active biochemical systems, and chemical inertness. Moreover, their surface chemistry enables the functionalization of these carbon platforms via strong covalent or noncovalent methods with surface modifiers, which improves their electrochemical performance [26][27][28,29]. The research interest on carbon for electrocatalysis is also stimulated by the need to develop efficient electrodes for energy utilization (from fuel cells to batteries, photoanodes, and solar cells). In fact, to meet the demanding expectation for more sustainable and efficient conversion and storage of energy it is necessary to give proper attention to the conductive properties of (some) carbon materials and the possibility of fine tuning their nanostructures[28][29] [30,31]. The recent recognition that carbon materials used in electrochemical devices exhibit catalytic behavior in addition to electrochemical properties has moved many research groups working in catalysis to the electrochemical field bringing back new expertise on catalysis.
2. Aluminium Smelter Technology
For the reasons mentioned above and others, the aplication of advanced carbon-based materials was fast growing over the last decade. As a matter of fact, there was an exponential increase in the field of carbon and catalysis, particularly in nano and electrocatalytic aspects during the decade[30][31][32][33][34][35] [32–37]. This was the motivation to contribute a communication to the Symposium ‘Carbocat VIII’ directed particularly at the carbon anode and its traditional and new possibilities for the 21st century. More specifically, this article examines, briefly, how the several major industries, associated with carbon artifact production, essentially belong to one, closely knit family, whose common parents are the geological fossils called petroleum and coal and, also, attempts to review some important applications of carbon electrodes, with a major focus on anode electrocatalysts developed over the last 30 years or so. In this context, the article`s introduction is followed by an account of structure in carbons and carbon forms, followed by catalysis of carbon oxidation reactions, nanotechnology, and carbon electrocatalysis. Then, it deals with several applications of carbon anode electrodes that are summarized here, beginning with the aluminium production.
Aluminium is the most abundant metal in the Earth’s crust, and the third most abundant element, after oxygen and silicon. It makes up about 8% by weight of the Earth’s solid surface. Rapid dissolution of aluminium in any acid and alkaline solutions is always found, but around neutral pH, the oxide formed is very dense and non-conducting, and oxidation is effectively stopped after a thin layer of about 5 nm has been formed. This thin layer of oxide permits aluminium to be used as a construction material and in many other day-to-day applications.
Since aluminium is quite an active metal, the traditional smelting technique used for iron did not work, and electrolysis, with the significant development of the electrical generator, was the only practical method to enable the electrolytic aluminium production. Reduction of Al (III) from aqueous solution was also impossible since hydrogen would be evolved first even from strongly basic solutions. The solution to these restrictions was discovered in 1886 independently by Hall in the United States, and Héroult in France.
Considering the cathode and anode primary reactions, in an aluminium cell, which can be simplified as:
|
|
(1) |
|
|
(2) |
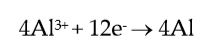
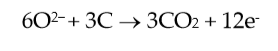
the overall cell reaction can be written as:
|
|
(3) |
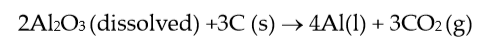
A typical aluminium reduction cell is shown in Figure 2.
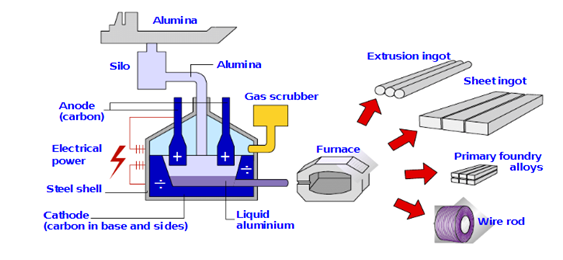
Figure 2. Scheme of aluminium production cell.
The total cell potential in the Hall-Héroult process is comprised of three different contributions, which are: (i) The reversible cell potential for the overall reaction (influenced by cell temperature and alumina concentration); (ii) the electrode overpotentials required for the occurrence of the anode and the catode partial reactions at a reasonable rate (depends on operating conditions); and (iii) the ohmic drop (due to the resistance in the electrolyte and in the electrodes). The reversible cell potential is about −1.215 V; for an ideal cell process, and since 3 Faradays of charge are required per mole of the aluminium, we would expect an electric power consumption of about 350 kJ/mol. The ohmic drop in the electrolyte is about −1.535 V, a large value that may be due to the large interelectrode gap, and the undissolved alumina. The ohmic drop in the anode is about −0.420 V, and in the cathode is about −0.680 V, values that may be ascribed to the low carbon conductivity, gas bubbles on the anode, and impurities such as P and V, which show variable oxidation states, being reduced at the cathode and then reoxidized at the anode, consuming current without production any metal. The anode and cathode overpotentials are about −0.510 and −0.080 V, respectively. Thus, the total working cell potential is about −4.450 V. Added to this is the energy cost of heating the cell to 1000 °C, purifying and drying the bauxite, and preparing the carbon anodes. A typical cell size was about 10–20 kA in 1914, and 50 kA in 1940. Today, new cells operating at 400 kA are quite normal, and some companies are now working with concepts to reach above 600 kA. A typical cell house will contain about 400 cells arranged in series on two lines, with each 6 × 8 m cell having a total anode area of 30 square meter. The optimum current density is around 1 A cm−2, giving a total cell current of 300 kA, and this requires a cell potential in the range from −4.0 to −4.5 V. The cell potential, of course, depends on alumina concentration since this determines the concentration of electroactive species at both electrodes. It drops to just below −4.0 V after addition of alumina to 6% and rises to about −4.5 V before the onset of an anode effect. Hence, the cells are arranged in the cell house to produce the minimum magnetic field. The primary anode reaction always leads to some loss in current efficiency, and in most cells, the aluminium current efficiency is only 85–90%. From these data, the energy requirement may be estimated to be over 14,000–16,000 kWh per ton of aluminium, and we can also calculate that the cell house described would produce 140,000 ton/year.
3. Electrochemical Kinetics
More than ever before, electrochemists now address problems of general scientific interest and use a large variety of other techniques, while researchers in many fields routinely resort to electrochemical measurements to obtain essential information. Consequently, electrochemistry has become an important facet of modern science, especially in surface and materials science. In this context, the basic physical principles that govern electrochemical systems, including bulk electrochemistry and interfacial electrochemistry, have been intensively researched during the last two decades. This is particularly obvious in the case of electrocatalysis with the aim of increasing the performance of novel electrocatalysts for a required carbon electrode process. By using different types of carbons (glassy carbon, carbon felt, carbon paper, graphite, graphite-reinforced carbon, carbon fibers, reticulated vitreous carbon, carbon nanowires, carbon nanotubes, carbon dots, graphene flakes, graphene/graphene oxide foams, carbon decorated with nanoparticles, doped with heteroatoms, etc.), prepared or modified as several reported above, it has been possible to obtain significant information about current relevant electrode processes[36][37][38][39][40][41][42][43][44][45][46][47][48][49][50] [154–168]. In this context, here we will discuss some emerging heterogeneous electrocatalysts and relevant electrochemical transformations involving water, hydrogen, oxygen, hydrogen peroxide, carbon dioxide, and nitrogen, whose address would allow the sustainable production of many fuels and chemicals.
3.1. Hydrogen Reactions
The electrolytic hydrogen technology has been used for industrial production of hydrogen for more than 100 years, and one of the most studied problems in electrode kinetics is the reduction of hydrogen ions to form hydrogen gas[51][52][53][54][55][56] [169–174]. One of the most striking features of the experimental results is the enormous range in the exchange current density with electrode material. Hydrogen evolution at a platinum electode has an extremely high exchange current density when compared with its value at a lead electrode. Furthermore, the apparent cathode transfer coefficients vary from less than 0.5 to about 2.0. These features can be understood, at least qualitatively, in terms of a simple model, as described below. An advanced catalyst for electrochemical hydrogen evolution reaction (HER; 2H+ + 2e−→− ® H2) should reduce the overpotential, and consequently increase the efficiency of this important electrochemical process[56] [174]. The HER is a classic example of a 2-electron transfer reaction with 1 catalytic intermediate, H*, where * denotes a site on the electrode surface. In electrode kinetics, its reaction rate vs. overpotential relationship can be expressed by the Butler-Volmer equation[57][58] [76,77], which can be simplified to the well-known empirical Tafel relation for overpotentials higher than 50 mV. Then, two important parameters (the Tafel coefficient, b, and the exchange current density, j0) can be obtained in which the dominant reaction mechanism of HER processes can be revealed based on the value of b. In general, for the HER under acidic conditions, at room temperature and 1.0 atm., H+ in the electrolyte obtains an electron and subsequently forms an adsorbed hydrogen atom on the active site of the electrocatalyst; this is referred to as the Volmer reaction (Volmer step 4). As for the subsequent reaction step involving the desorption process, this can proceed through two different mechanisms to produce hydrogen. In the case, in which the adsorbed hydrogen atom coverage is relatively low, the adsorbed species prefers to react with H+ and electrons to evolve H2 through an electrochemical desorption process that is referred to as the Heyrovsky reaction (Heyrovsky step 5). As the H* coverage is relatively high during the process of reaction, the dominant desorption processes will be transferred from above electrochemical desorption to recombination process between adjacent H*, which is also referred to as a Tafel reaction or a chemical desorption reaction (Tafel step 6). If b is −30 mV/dec, it means that step 4 is fast, and the chemical desorption process is the rate-determining step (Tafel step 6). If b is −40 mV/dec, this also indicates that step 4 is fast, but that the hydrogen desorption rate is relatively slow and therefore hydrogen is evolved through the electrochemical desorption process as the rate-determining step (Heyrovsky step 5). Lastly, in cases in which b is −120 mV/dec, this indicates that the first step is slow (Volmer step 4) regardless of whether hydrogen is evolved through the electrochemical desorption reaction or the chemical desorption reaction. As for the value of j0, this is another important parameter to evaluate the intrinsic activity of electrocatalysts in which the electrochemical reaction rate at reversible conditions can be obtained. In general, small b and large j0 values are desirable for ideal electrocatalysts. In summary, the HER may occur through either the Volmer-Heyrovsky or the Volmer-Tafel mechanism[59][60][61] [83,175,176].
|
|
(4) |
|
|
(5) |
|
|
(6) |
Tafel slope, b, is the inherent property of a catalyst, and is determined by the rate limiting step of HER; the b values quoted are for 25 °C and a symmetry coefficient of 0.5[60] [175]. As for alkaline conditions, the adsorbed hydrogen atom is generated from the electrochemical reduction of H2O, which is more sluggish than the reduction of H+ in acidic conditions because the H-O-H bonds need to be broken before adsorbing hydrogen. Therefore, it is more facile to evolve hydrogen in acidic solutions than in alkaline soluions.
3.2. Oxygen Reactions
Oxygen evolution reaction (OER) is a crucial reaction for many energy technologies such as high efficiency water electrolyzers, or photo-driven water splitting, regenerative fuel cells, and advanced rechargeable metal-air batteries. Accordingly, high performance catalysts are urgently needed to speed up the OER, lower the high overpotential required to drive the reaction and reduce the energy consumption.
Two aspects of the OER are common to all electrodes studied so far: (i) The exchange-current density is low, of the order of 10−10 A/cm2 or less, and (ii) a reversible oxygen electrode operating at or near room temperature has not yet been found. At sufficiently high temperatures (say, in molten salts, at about 600 °C, or with high temperature solid electrolytes operating at around 1000 °C) the kinetics of the reaction can be sufficiently accelerated to make reversible oxygen electrodes operate.
An important point that is often ignored is that by the time the reversible potential for oxygen evolution is reached, an oxide layer has been formed on all metals. At more anodic potentials, where measurements can actually be conducted to follow the oxygen evolution, the oxide film may be several molecular layers thick. In other words, the OER never occurs on the bare metal surface.
Oxygen evolution can occur readily on the electronically conducting oxides such as ruthenium dioxide and the oxides formed on platinum and iridium. On semiconducting oxides such as NiO, and the oxides formed on tungsten and molybdenum, the OER can still occur, but it may be associated with pitting on the one hand and further build up of the oxide layer on the other, causing poor reproducibility and making the interpretation rather dubious. On insulators, such as alumina and the oxides formed on the valve metals (Ti, Ta, and Nb), oxide formation is the main reaction occurring during anodic polarization and the current either decays to zero with time or reaches a constant value, at which the rate of dissolution of the oxide is equal to the rate of its electrochemical formation.
On noble metals (e.g., Pt, Ir, Au), the region of potential in which the OER is studied is far removed from the potential of zero charge in the positive direction. Thus, only negatively charged impurities will be heavily adsorbed, and the nature of the anion in the electrolyte will influence the measurements substantially. Also, most organic molecules are rapidly oxidized at these potentials, so that the requirements for solution purification are much less severe than in the case of hydrogen evolutio
In almost all applications of the oxygen reduction reaction (but not the hydrogen peroxide production), it is desirable to select conditions where the complete 4-electron reduction reaction (ORR; O2 + 4H+ + 4e− → ® 2H2O) occurs; i.e., in acid solution (pH 0, ORR formal potential +1.23 V vs. SHE) or in basic solution (pH 14, ORR formal potential +0.39 V vs. SHE). In any case, the oxygen electrode is a complex system and the overall reaction in either direction requires the transfer of four electrons and four protons. The majority of the research on the ORR has been centered on the use of noble metal electrodes, due to their relative stability in acidic or alkaline solutions. The preceding statement on noble metals for the OER does not hold true for the ORR. Using platinum as an example, we note that oxygen evolution is typically studied in the range 1.5–2.0 V vs. a reversible hydrogen electrode (RHE). In the same solution, oxygen reduction is studied in the range 1.0–0.4 V on the same scale. In most of the latter range, the surface is free of oxide (and very sensitive to impurities) if approached from low potentials, whereas it may be covered partially with oxide (and less sensitive to impurities) when approached from higher potentials. This is due to the high degree of irreversibility of formation and removal of the oxide layer on most noble metals. Again, considering statements above about the OER, it should be noted that the ORR may well occur on the bare metal surface, or one that is covered only by a fraction of a monolayer. Thus, even when Pt is used, it is well worth to remember that the oxidation and reduction of oxygen on the same metal occur at different surfaces and may therefore follow entirely different pathways.
3.3. Hydrogen Peroxide Production
Hydrogen peroxide, well known for the last 200 years, is a very clean chemical with high versatility. It finds enormous application as a bleaching agent in the pulp, paper, and textile industries, as well as in the cosmetics and medicinal fields and the food processing industry. It is also used as an oxygen source and as an oxidizing agent in the mining and electronic industries. Being environmentally and ecologically friendly finds use in a variety of applications related to the environment. Highly concentrated hydrogen peroxide also finds use as a propellant iin the transportation industry.
The cathodic reduction of dissolved oxygen to peroxide was first demonstrated by Traube in 1882, but only over the last 50 years, there has been a gradual awareness of the desirable factors for a successful process, and cells developed by the Dow Chemical Company and others, lead to developed hydrogen peroxide production systems.
Overall, the production of hydrogen peroxide from oxygen involves two coupled electron-proton transfers and one reaction intermediate (OOH*), making it similar in complexity to the HER[62] [220].
|
|
(7) |
|
|
(8) |
As such, it is possible to find a catalyst with zero theoretical overpotential that has an optimal DGOOH, binding OOH* neither too strongly nor weakly[62] [220]. Pt[63] [249], Ag[64] [250], Au[65] [251], Au-Pd alloys[66] [252], nitrogen-doped carbon[67] [253], and hierarchically porous carbon[68] [254] have been explored for the hydrogen peroxide production, but their activities and selectivities are very modest.
3.4. Carbon Dioxide Reduction Reaction
The possibility of capture and storage of carbon dioxide in various media like amines, zeolites, and metal organic frameworks, as well as in geological systems, oceans, and by mineral carbonation has been technologically considered[69][70][71] [273–275]. The capture and storage of carbon dioxide emissions can also be considered as a valuable resource because CO2 can be catalytically converted into industrially relevant chemicals and fuels[72][73][74] [80,81,276]. A proposed method for the conversion of CO2 to value-added chemicals is the carbon dioxide reduction reaction (CRR), which was investigated on a wide variety of hererogeneous elemental surfaces, consisting of metal electrodes and other electrodes including metal complexes[74] [277]. The CRR uses environmentally benign aqueous electrolytes, easily couples with electricity sources, and the reaction rate can be controlled easily by tuning the external bias (i.e., the overpotential). However, the currently known catalysts are very limited in terms of overpotential, selectivity, production rate, activity, and durability, hampering this process from becoming close to commercialization.
Linear carbon dioxide molecule is a non-polar, fully oxidized, and extremely stable molecule. To induce its chemical conversion, a commercial electrocatalyst requires a large overpotential to promote the conversion reaction at satisfactory rates and high product selectivity.
3.5. Nitrogen Reduction Reaction
Nitrogen is one of the most abundant elements in the atmosphere and most inert also. The electrochemical reduction of nitrogen (NRR) is more difficult than the CRR due to presence of three covalent bond between nitrogen atoms, which have high bond energy of 941 kJ/mol and therefore difficult to perform. For the NRR suitable catalysts are required, and Earth Abundant Electrocatalysts (EAEs) are good candidates for this reduction.
The most accepted mechanism of NRR contains associative and dissociative paths. Firstly, the nitrogen molecules are adsorbed on the catalyst surface and then the hydrogenation process proceeds. As with CRR, the NRR involves multiple intermediates, and the HER is a major competing reaction, making selectivity a great challenge[75] [287].
The electrochemical N2-to-NH3 process is currently being actively studied due to the importance of ammonia as fertilizer and in several plastics, fibers, explosives, pharmaceutical and textile industrial applications, as well as a high viable chemical energy carrier. The fundamental issue in much of the research on this electrochemical NRR process to form ammonia from nitrogen and water, is that the quantities of ammonia produced are very small. To minimize this problem, and in particular the simultaneous reduction of nitrogen and hydrogen, inhibiting the HER process, electrocatalysts are the paramount components. Thus, it is a priority to synthesize NRR electrocatalysts and rationally design them to optimize the mass transport, chemi (physi)sorption, and transfer of protons and electrons. Three important figures of merit are also being considered: The selectively of NRR over the HER, the energy efficiency of the overall process, and the system throughput of NH3 synthesis. Model systems in the nano-aqueous environment offer a guide to understanding the fundamental proton-coupled electron transfer (PCET) requirements needed to effect NRR over HER[76][77][78][79][80][81][82][83] [289–296]. The parameter space for advanced catalytic molecules and materials remains to be explored. In this regard, computational studies help facilitate the catalyst design[84] [288], while fine-tuning the active sites at nanoscale with chemical insight will be welcomed as a beneficial degree of freedom[85] [297]. Conversely, hybrid biological inorganic (HBI) approaches[86][87][88] [298–300] provide selectivity at ambient conditions and promise high-energy efficiencies, but with the challenge of sufficient throughput. Regardless of the recent use of sophisticated techniques and appropriate detection method protocols, the exact mechanism pathways on any given metal surface or flat and stepped surfaces (e.g., of noble metal, non-noble metal, and metal-free electrocatalysts) remains elusive for this hydrogen vs. ammonia issue.
3.6. Borohydride Oxidation Reaction
The Brown–Schlesinger and the Bayer processes were the first ways for producing NaBH4[89][90][91] [301–303]. Sodium borohydride is non-toxic, easily stored, has a high energy density (9.3 Wg−1), can be massively produced from trimethyl borate and borax, has long-term stability (half-life 426 days at pH 12), has potential usage at ambient conditions, has high hydrogen storage content (10.6 wt.%), releases up to eight electrons at an electrode potential as low as −1.24 V vs. SHE, and is pollution-free (the borohydide oxidation reaction, BOR, does not release carbon and nitrogen compounds, e.g., carbon dioxide, nitrogen dioxide, and so on). Apart from its oxidation, it has become the focus of many studies particularly in the field of the borohydride fuel cells[92] [304]. The key factor for practical application of direct borohydride fuel cells (DBFCs) is to prepare anode electrodes (catalytic or not with respect to the borohydride hydrolysis) that have high selectivity and high catalytic activity for improving the kinetic parameters of BH4- oxidation and inhibiting hydrolysis of BH4−. The ideal eight-electron BOR process can be understood as the oxidation of all the H(−1) in the borohydride anion into H(+1) in water. However, some of the H will be lost in the form of H2 during the oxidation, resulting in incomplete utilization of the eight electrons. The loss of faradaic efficiency due to the HER is a major issue limiting the performance of DBFCs. From the point of view of the cathode catalysts for the oxygen electrocatalysts, the alkaline electrolyte needed to stabilize the borohydride offers the possibility of using precious metals, non-PGM materials, activated and metal-doped carbons, nonmetal-doped carbon, carbon-transition metal hydrides, transition metal oxides with spinel and perovskite structures, and so forth. For alkaline fuel cells using carbon catalysts, the goal is to modify the cost-effective carbon-based electrocatalysts to increase the number of electrons up to four and to reduce the cathode activation overpotential. Pt-free ORR catalysts, the transition metal, nitrogen, and carbon groups, or M-N-C materials, are attractive candidates due to their high surface area, high activity, and low cost. The M-N-C synthesis involves various precursor deposition steps onto the high surface area carbons. The final catalysts consist of a combination of the active material with an inert carbon matrix, which substantially decreases the density of active sites for the four-electron pathway.
4. Direct Carbon Fuel Cell
Carbon materials and carbon nanomaterials are applied in many fuel cell technologies[93][94][95][96][97] [319–323], which are being extensively explored. On the other hand, in the direct carbon fuel cell (DCFC), the overall investment is relatively small, and considerable effort is required to take this technology to the pre-commercialization stage. For this reason, and the fact that carbons, namely carbon anodes, play a key role in this system, we have decided to analyse this fuel cell here.
In a DCFC, each cell consists of a cathode and anode separated by an ionically conducting but electronically insulating electrolyte. Moreover, the anodic compartment is supplied with a solid fuel that reacts directly at the electrode/electrolyte interface or line of contact where the reactant, electronic conductor, and ionically conductive phase all meet, to form a gaseous exhaust product. These interfaces are known as triple-phase boundaries (TPBs). In the DCFC, the chemical energy in the carbon fuel (turned into submicron size particles) is directly converted into electricity, the overall cell reaction occurring at 500–900 °C:
|
|
(9) |
|
|
(10) |
Mostly, the developmental work on this technology has so far been concentrated on button cells of ceramic electrolyte disk with nickel-based anode and lanthanum strontium manganite (LSM) based cathode [98][99][327,328]. The major technical issues apart from those associated with SOFC are the solid fuel delivery to anode/electrolyte interface, and the lack of understanding of carbon oxidation reaction mechanisms at the interface.
5. Neurochemical Monitoring
Microelectrodes, i.e., those with radii on the order of a few micrometers, have many unusual advantages over large electrodes, which are of interest to researchers in the field of sensor development. Because of the small size of microelectrodes, the absolute magnitude of the current obtained during an electrochemical experiment is quite small, usually a few hundred picoamps to several nanoamps. Currents of this magnitude are advantageous for the elimination of distortion in voltammetric response curves arising from the ohmic potential drop between the working and reference electrodes. This has led to the application of electrodes of this type to electroanalysis in non-polar organic solvents as well as in the absence of added supporting electrolyte. In situ monitoring in flow streams of changing ionic strength with microelectrodes, for example, should therefore be less prone to distortions in response due to changing resistivity of the bulk medium.
More recently, it has been possible to design microelectrodes for biomedical applications of even smaller dimensions and lower electrical resistance in which the electronic conductor is a carbon fiber. Carbon fiber electrodes (CFEs) were first invented in the late 1970’s by François Gonon and colleagues[100][101] [329,330], who discovered the carbon fiber electrode`s abilities to qualitatively detect species in the inner working of a brain by electrochemical pretreatment of the CFE before implantation[102] [331].
Previous preliminary studies showed that neurotransmitters and their metabolites were merely a percentage of key redox species present in the brain. In the 1980s, Mark Wightman and colleagues[103] [332] determined guidelines to detect and identify in vivo species, which are now well known as the ‘Five Golden Rules. These golden rules are discussed in a book on voltammetry in the neurosciences[104] [333] and the journal Trends in Analytical Chemistry[105] [334].
Microelectrodes come in all shapes and sizes[106] [109], being capable for micro- measuring devices (Figure 3) [107][337].
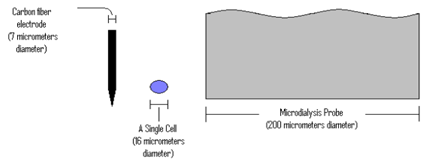
Figure 3. The relative sizes of a carbon fiber electrode and a microdialysis probe next to a single cell.
Future directions for the carbon microelectrode are likely related to its size. At present, carbon nanotubes (CNTs) are the point of interest[108][109][110] [346–348]. Apart from their use in neurochemical monitoring, carbon fibers and carbon nanotubes, either single-walled SWCNTs or multi-walled MWCNTs[111][112][113] [349–351], are finding their way in different applications that touch nearly every field of technology including aerospace[114] [352], electronics[115] [353], medicine[116] [354], defense [117][355], automotive[117] [355], energy[118] [356], construction[119] [357], and even fashion[120] [358]. In particular, the ability of CNTs to effectively target the cancer cells may revolutionize our approaches in treating this dreaded disease and bring us near the Holy Grail[121][122][123][124][125][126][127] [359–365].
6. Lithium Ion Batteries
Lithium batteries were first proposed, by M. S. Whittingham around the 1970s[128] [383]. The group IV and V dichalcogenides, later identified as intercalation compounds, attracted attention for their high electrical conductivities, and the fact that they react with alkali metals in a reversible way. In 1972, Exxon[129] [384] initiated a large project on solid state batteries, using TiS2 as the positive electrode, Li metal as the negative electrode and lithium perchlorate in dioxolane as the electrolyte. Titanium disulfide was chosen as a cathode material because it is the lightest and cheapest of all groups IV and V layered dichalcogenides and its ability to undergo intercalation upon treatment with electropositive elements. TiS2 adopts a hexagonal close packed structure, each sulfide being connected to three Ti centers, the geometry at S being pyramidal[130] [385]. The individual layers of TiS2, which consist of Ti-S bonds, are bounded together by relatively weak intermolecular van der Waals forces. Unfortunately, it was soon realized that using lithium as an anode material lower the performance of the batteries (low capacity and cycle life) and made them unsafe due to dendritic Li growth during charge-discharge cycling. These dendrites or pesky formations are the reasons that most lithium batteries on the market today are lithium-ion rather than lithium-metal.
The 1979 discovery at Oxford University that Li+ ions may be electrochemically withdrawn from the LiCoO2 and LiNiO2 structures and replaced reversibly, that is, either of these compounds can be used as the active material for a positive electrode in a 4 V rechargeable lithium cell, was a significant advance. In 1990, the Sony Corporation in Japan, announced the <lithium-ion battery>, that was the first rechargeable lithium battery depending entirely on the difference in electrochemical potential of lithium ions intercalated in LiCoO2 and graphite.
To obtain high energy density batteries, graphite, graphitizable carbon (soft carbon), and nongraphitizable carbon (hard carbon)[131] [386], with large doping capacities, and the possibility of lithium-carbon intercalation complexes exceeding the LiC6 stoichiometric composition, are being studied and employed as anodes.
One great challenge in the development of lithium ion batteries is to simultaneously achieve high power and large energy capacity at fast charge and discharge rates for several minutes to seconds[132] [414]. In this aspect, transition metal oxides, silicon, tin, and zinc, etc., with addition of additives to mitigate volume changes observed during cycling have been explored as active anode materials to replace graphite because of their high theoretical capacities.
In recent years, graphene has been employed as an encapsulating agent for these materials. So, this work that eventually provides materials with high capacities requires consideration. Furthermore, stacked sheets of graphene derived from exfoliated graphite provide a modular approach to exploring lithium storage in layered carbon as well as layered carbon/metal nanocomposite[132] [415].
Owing to its high theoretical capacity (4200 mAh/g) when alloying with lithium, and the fact that is the second most abundant element on earth, silicon has received a huge attention as a prospective replacement material for use as anodes in a LIB[133][134] [60,416] though it is actually a metal. The alloying of silicon with lithium is associated with a volume expansion of more than 300%, leading to pulverization, which results in loss of electrical contact and eventual fading of capacity. Enormous efforts have been made to overcome these problems by using nanostructured Si materials and others, but it remains challenging via facile approaches to achieve long cycle life and high capacity of Si anode materials in large scale[135][136][137][138][139] [417–421].
Further studies[140][141][142] [429–431] led to three-dimensional (3D) silicon/carbon/graphene nano composites, three-dimensional (3D) graphene-carbon nanotube-metal/metal oxide nanocomposites, three-dimensional (3D) graphene-carbon nanotube–TiO2 nanocomposites, three-dimensional (3D) graphene-carbon nanotube-nickel nanocomposites, and other novel 3D functional nanostructures, namely three-dimensional (3D) graphene-nanotube-iron hierarchical nanocomposites and doped hierarchical porous graphene electrodes[143][144][145][146] [432–435] with outstanding structural properties, minimum contact resistance, enhanced Li-ion diffusion, high capacity, high energy density, and prevention of the restacking of graphene layers. In addition, these structures overcome the agglomeration of metal/metal oxides nanoparticles in the case of 3D composites.
7. Electrochemical Capacitors
Renewable energies, that is, natural energy resources such as wind power, solar energy, and geothermal energy, are converted to usable energy, mainly electric energy, which is stored in batteries and capacitors, particularly of the type’s lithium-ion and electrochemical capacitor. LIBs were analysed in the previous section. Here, we deal essentially with electrochemical double layer capacitors (EDLCs). These batteries and capacitors utilize carbon materials as electrodes.
A conventional electrostatic capacitor consists of two metal plates of equal area, A, separated by an insulator (a dielectric, e.g., vacuum, air, mica, oil, paper, plastic). By means of an applied voltage, the device stores energy by the separation of positive and negative electrostatic charge, q, between the parallel plates. If the potential difference between the plates is Vc and the dielectric constant of the insulating material is , then the capacitance, C, is given by C = q/Vc = © A/d, where d is the inter-plate spacing. The energy stored in a charge capacitor is given by U = ½ C Vc2. The energy density is very low, of the order of 0.05 Wh dm−3.
In an electrolytic capacitor, the dielectric is a very thin oxide film formed electrolytically on a metal such as Al, Ta, Ti, or Nb. The metal substrate acts as one conducting phase and an electrolyte solution as the other. Given that the dielectric is extremely thin, the specific capacitances (F/g) of electrolytic capacitors are much larger (up to 1000 times) than those of electrostatic counterparts. The specific energy is higher (typically 0.06 vs. 0.003 Wh/kg), but both types of capacitor are extremely poor energy-storage devices.
So-called supercapacitors store electrostatic charge in the form of ions, rather than electrons, on the surfaces of materials with high specific areas (m2/g). The electrodes are of finely divided, porous carbon, which provide a great charge density. The voltage is lower than for a conventional capacitor, while the time for charge-discharge is longer because ions move and reorientate more slowly than electrons.
By combining an electrode material with a large specific surface area with a material that can be reversibly oxidized and reduced over a wide potential range, it is possible to realise a device close to a battery, that is the ultracapacitor. The energy is stored both by ionic capacitance and by surface (and near surface) redox processes that occur during charge and discharge. This enhances the amount of stored energy. Moreover, because the ions are confined to surface layers, the redox reactions are rapid and are fully reversible many thousands of times, which therefore make for a long lifecycle.
Electrochemical capacitors (ECs) can store vastly more energy than conventional capacitors. They vary in size from small capacitors used in electronics to devices with capacities >3000 F that form the basic module for the units used in hybrid electric vehicles. They may be discharged at rates up to 10 to 25 times faster than batteries and, equally importantly, can also be recharged at much greater rates than batteries. Moreover, they have very long lives and operate satisfactorily at temperatures as low as −40 °C.
Electrical energy storage in ECs occurs due to the formation of electric double-layer (EDL) on the electrodes’ surfaces and to some extent surface oxidation/reduction. The capacitance due to the former (EDLC) is called electric double-layer capacitance and that due to the latter is called pseudo-capacitante. The capacitors that consist of different mechanisms, for example, intercalation/deintercalation and adsorption/desorption, are called hybrid capacitors. Activated carbons with a large surface area, good electric conductivity, electrochemical inertness, and lightweight properties, are excellent materials to increase the capacitance, so that they are very employed as carbon electrodes. Many reviews and articles on carbon materials to EDLCs have been published[147][148][149][150][151] [436–440].
Recently, hybrid energy storage devices termed as supercapattery, are developed to complement the figures of supercapacitors and batteries. One supercapattery uses redox active battery grade materials as positive electrode with the high power delivery capability and carbonaceous materials as negative electrode I 512 I. Thus, complementing the advantages of both batteries as well as supercapacitors, i.e., possessing energy as much as the battery and high power output almost as much as the supercapacitor.
8. Conclusions
Looking back at the carbon highlights reported here, we clearly found some areas deserving attention. It is the case of the aluminium production in large alumina refineries, using carbon anodes of high quality, which depends on the characteristics of coke filler, coal tar pitch binder, and anode scrap, among others. However, then we can see the development of synthetic diamonds by the GE high pressure catalytic process initiated in 1941, leading to the first commercially successful synthesis on december 1954. Much later the diamond and diamond-like films appeared, using low temperatures and low-pressure procedures, truly defiant of all the laws of thermodynamics and phase diagrams. The carbon fibers are other excellent carbon materials whose development led to the carbon fiber reinforced plastic (CFRP) and other composite products, which have several uses in aerospace and non-aerospace structures, as well as in non-structural applications. One of the most intriguing discovery has been the fullerene systems and the nanotubes that are capturing the imagination of physicists, chemists, materials scientists, and nanotechnologists alike. These new discoveries and developments had an impact that extended well beyond the confines of academic research and marked the beginning of a new era in carbon science and technology, namely in the field of carbon electrochemistry.
Another factor stimulating the research interest on carbon for electrocatalysis is the worldwide need to develop more efficient approaches for electrode materials for more sustainable utilization of energy together with the possibility of fine tuning of their nanostructure to realize advanced electrodes to meet the demanding expectation for more sustainable and efficient conversion and storage of energy.
For a better understanding of these carbon-based electrodes development and applications, this article includes a brief account of structure in carbons and carbon forms, followed by catalysis of carbon oxidation reactions, nanotechnology, and carbon electrocatalysis. The traditional aluminium electrolysis is then reported, followed by the application of carbon materials in electrochemical kinetics and new electrochemical energy technologies, namely direct carbon fuel cells, lithium ion batteries, and electrochemical capacitors. Updated applications such as neurochemical monitoring and supercapattery are also reported.