Prostate cancer is the second most frequent cancer in males (15.1%) and the fifth cause of cancer-related death (6.83%) in men worldwide. Although the traditional treatment approach, which includes surgical resection, radiotherapy, and hormone therapy, has led to great improvements in both survival and quality of life in patients with localized disease, the prognosis for metastatic disease remains poor. Molecular targeted therapies, aimed at blocking specific molecules or signaling pathways in tumor cells or in their microenvironment, with a low risk of damage to normal tissues, have demonstrated their efficacy in several types of cancer. Various trials have had encouraging results in the treatment of metastatic prostate cancer.
- anti-angiogenic therapy
- castration-resistant prostate cancer
- immunotherapy
- metastasis
- molecular targeted therapies
1. Introduction
In Europe, prostate cancer is the most frequent malignant tumor (22.2%) and the third cause of cancer-related death (10.0%) in men, after lung and colorectal cancers [1].
Since ~65% of new prostate cancers are diagnosed in males over 65 years old and in 25% of males over 75 years old, the incidence of prostate cancer and related mortality are expected to rise due to an increase in life expectancy and the aging population. Therefore, there is a critical need for the development of innovative and tolerable therapeutic approaches, effective in the treatment of advanced disease and suitable for frail elderly patients.
Molecular targeted therapies, aimed at blocking specific molecules or signaling pathways in tumor cells or in their microenvironment, with a low risk of damage to normal tissues, have demonstrated their efficacy in several types of cancer. Various trials have had encouraging results in the treatment of metastatic prostate cancer.
The molecular targeted therapies under study, or that have entered clinical use or trials for the treatment of advanced prostate cancer, belong to one of four categories: prostate-specific membrane an-tigen (PSMA)-targeted radionuclide therapies; DNA repair inhibitors; therapies targeting tumor neovascularization; or immune checkpoint inhibitors.
2. PSMA-Targeted Radionuclide Therapies
3. DNA Repair Inhibitors
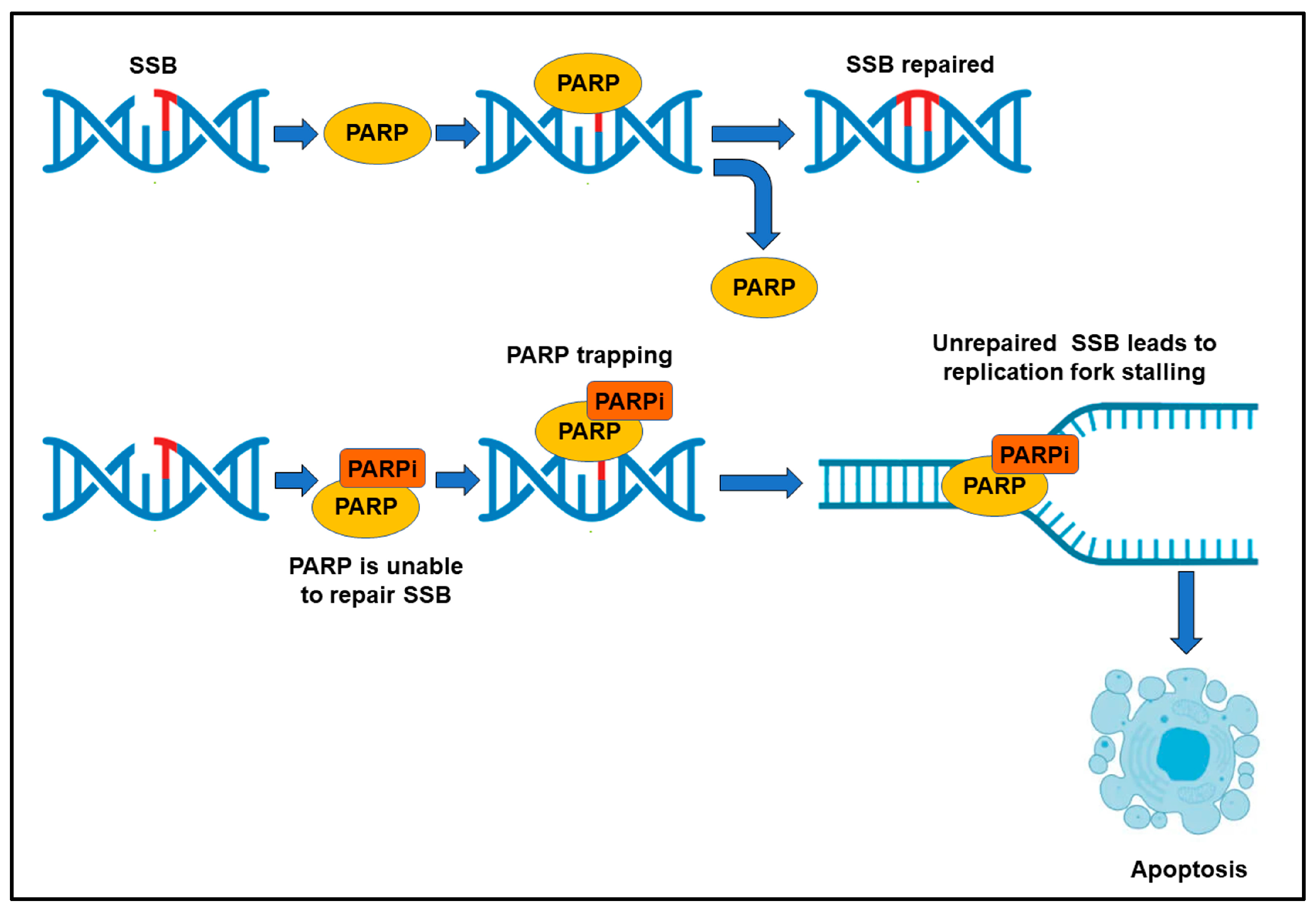
4. Therapies Targeting Tumor Neovascularization
5. Immune Checkpoint Inhibitors
References
- Global Cancer Observatory. Available online: https://gco.iarc.fr/ (accessed on 19 May 2023).
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608.
- Porter, A.; McEwan, A.; Powe, J.; Reid, R.; McGowan, D.; Lukka, H.; Sathyanarayana, J.; Yakemchuk, V.; Thomas, G.; Erlich, L.; et al. Results of a randomized phase-III trial to evaluate the efficacy of strontium-89 adjuvant to local field external beam irradiation in the management of endocrine resistant metastatic prostate cancer. Int. J. Radiat. Oncol. 1993, 25, 805–813.
- Lepor, H. Management of clinically localized prostate cancer. Rev. Urol. 2004, 6, S3–S12.
- Chang, S.S. Overview of Prostate-Specific Membrane Antigen. Rev. Urol. 2004, 6 (Suppl. 10), S13–S18.
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177–PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103.
- Hofman, M.S.; Emmett, L.; Sandhu, S.; Iravani, A.; Joshua, A.M.; Goh, J.C.; Pattison, D.A.; Tan, T.H.; Kirkwood, I.D.; Francis, R.J.; et al. TheraP: 177Lu-PSMA-617 (LuPSMA) versus cabazitaxel in metastatic castration-resistant prostate cancer (mCRPC) progressing after docetaxel—Overall survival after median follow-up of 3 years (ANZUP 1603). J. Clin. Oncol. 2022, 40, 5000.
- El Fakiri, M.; Geis, N.M.; Ayada, N.; Eder, M.; Eder, A.-C. PSMA-Targeting Radiopharmaceuticals for Prostate Cancer Therapy: Recent Developments and Future Perspectives. Cancers 2021, 13, 3967.
- Kratochwil, C.; Haberkorn, U.; Giesel, F.L. Radionuclide Therapy of Metastatic Prostate Cancer. Semin. Nucl. Med. 2019, 49, 313–325.
- Featherstone, C.; Jackson, S.P. DNA double-strand break repair. Curr. Biol. 1999, 9, R759–R761.
- Liang, Y.; Lin, S.-Y.; Brunicardi, F.C.; Goss, J.; Li, K. DNA Damage Response Pathways in Tumor Suppression and Cancer Treatment. World J. Surg. 2008, 33, 661–666.
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263.
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393.
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228.
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453.
- Nombela, P.; Lozano, R.; Aytes, A.; Mateo, J.; Olmos, D.; Castro, E. BRCA2 and Other DDR Genes in Prostate Cancer. Cancers 2019, 11, 352.
- Davies, K.D.; Ng, T.L.; Estrada-Bernal, A.; Le, A.T.; Ennever, P.R.; Camidge, D.R.; Doebele, R.C.; Aisner, D.L. Dramatic Response to Crizotinib in a Patient With Lung Cancer Positive for an HLA-DRB1-MET Gene Fusion. JCO Precis. Oncol. 2017, 1, PO.17.00117.
- McGlynn, P.; Lloyd, R.G. Recombinational repair and restart of damaged replication forks. Nat. Rev. Mol. Cell Biol. 2002, 3, 859–870.
- Dasovich, M.; Beckett, M.Q.; Bailey, S.; Ong, S.-E.; Greenberg, M.M.; Leung, A.K.L. Identifying Poly(ADP-ribose)-Binding Proteins with Photoaffinity-Based Proteomics. J. Am. Chem. Soc. 2021, 143, 3037–3042.
- Krastev, D.B.; Pettitt, S.J.; Campbell, J.; Song, F.; Tanos, B.E.; Stoynov, S.S.; Ashworth, A.; Lord, C.J. Coupling bimolecular PARylation biosensors with genetic screens to identify PARylation targets. Nat. Commun. 2018, 9, 2016.
- Teloni, F.; Altmeyer, M. Readers of poly(ADP-ribose): Designed to be fit for purpose. Nucleic Acids Res. 2015, 44, 993–1006.
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158.
- Leung, D.K.W.; Chiu, P.K.F.; Ng, C.-F.; Teoh, J.Y.C. Novel Strategies for Treating Castration-Resistant Prostate Cancer. Biomedicines 2021, 9, 339.
- Antonarakis, E.S.; Wang, H.; Teply, B.A.; Kelly, W.K.; Willms, J.; Sullivan, R.; King, S.; Marshall, C.H.; Lotan, T. Interim results from a phase 2 study of olaparib (without ADT) in men with biochemically-recurrent prostate cancer after prostatectomy, with integrated biomarker analysis. J. Clin. Oncol. 2019, 37, 5045.
- Abida, W.; Campbell, D.; Patnaik, A.; Sautois, B.; Shapiro, J.; Vogelzang, N.; Bryce, A.; McDermott, R.; Ricci, F.; Rowe, J.; et al. Preliminary results from the TRITON2 study of rucaparib in patients (pts) with DNA damage repair (DDR)-deficient metastatic castration-resistant prostate cancer (mCRPC): Updated analyses. Ann. Oncol. 2019, 30, v327–v328.
- Markowski, M.C.; Wang, H.; Sullivan, R.; Haffner, M.; De Marzo, A.M.; Lotan, T.L.; Antonarakis, E.S. Phase II trial of rucaparib (Without ADT) in patients with metastatic hormone-sensitive prostate cancer harboring germline DNA repair gene mutations (TRIUMPH). J. Clin. Oncol. 2018, 36, TPS5095.
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763.
- Murai, J.; Huang, S.-Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599.
- Pommier, Y.; O’connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17.
- de Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): An open-label, phase 2 trial. Lancet Oncol. 2021, 22, 1250–1264.
- Teyssonneau, D.; Margot, H.; Cabart, M.; Anonnay, M.; Sargos, P.; Vuong, N.-S.; Soubeyran, I.; Sevenet, N.; Roubaud, G. Prostate cancer and PARP inhibitors: Progress and challenges. J. Hematol. Oncol. 2021, 14, 51.
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor–induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal. 2017, 10, eaam7479.
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanjiv, K.; Gelali, E.; Massie, C.E.; Baridi, A.; Warren, A.Y.; Zhao, W.; Ogris, C.; et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat. Commun. 2017, 8, 374.
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.-F.; Cai, L.; Zheng, D.; et al. Androgen Receptor Signaling Regulates DNA Repair in Prostate Cancers. Cancer Discov. 2013, 3, 1245–1253.
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual Roles of PARP-1 Promote Cancer Growth and Progression. Cancer Discov. 2012, 2, 1134–1149.
- Clarke, N.; Wiechno, P.; Alekseev, B.; Sala, N.; Jones, R.; Kocak, I.; Chiuri, V.E.; Jassem, J.; Fléchon, A.; Redfern, C.; et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018, 19, 975–986.
- Chi, K.N.; Rathkopf, D.E.; Smith, M.R.; Efstathiou, E.; Attard, G.; Olmos, D.; Lee, J.Y.; Small, E.J.; Gomes, A.J.; Roubaud, G.; et al. Phase 3 MAGNITUDE study: First results of niraparib (NIRA) with abiraterone acetate and prednisone (AAP) as first-line therapy in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) with and without homologous recombination repair (HRR) gene alterations. J. Clin. Oncol. 2022, 40, 12.
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882.
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676.
- Wong, S.Y.; Haack, H.; Crowley, D.; Barry, M.; Bronson, R.T.; Hynes, R.O. Tumor-Secreted Vascular Endothelial Growth Factor-C Is Necessary for Prostate Cancer Lymphangiogenesis, but Lymphangiogenesis Is Unnecessary for Lymph Node Metastasis. Cancer Res 2005, 65, 9789–9798.
- Wegiel, B.; Bjartell, A.; Ekberg, J.; Gadaleanu, V.; Brunhoff, C.; Persson, J.L. A role for cyclin A1 in mediating the autocrine expression of vascular endothelial growth factor in prostate cancer. Oncogene 2005, 24, 6385–6393.
- Green, M.M.; Hiley, C.T.; Shanks, J.H.; Bottomley, I.C.; West, C.M.; Cowan, R.A.; Stratford, I.J. Expression of vascular endothelial growth factor (VEGF) in locally invasive prostate cancer is prognostic for radiotherapy outcome. Int. J. Radiat. Oncol. 2007, 67, 84–90.
- Duque, J.L.F.; Loughlin, K.R.; Adam, R.M.; Kantoff, P.W.; Zurakowski, D.; Freeman, M.R. Plasma levels of vascular endothelial growth factor are increased in patients with metastatic prostate cancer. Urology 1999, 54, 523–527.
- Hrouda, D.; Nicol, D.; Gardiner, R. The role of angiogenesis in prostate development and the pathogenesis of prostate cancer. Urol. Res. 2003, 30, 347–355.
- Kelly, W.K.; Halabi, S.; Carducci, M.; George, D.; Mahoney, J.F.; Stadler, W.M.; Morris, M.; Kantoff, P.; Monk, J.P.; Kaplan, E.; et al. Randomized, Double-Blind, Placebo-Controlled Phase III Trial Comparing Docetaxel and Prednisone with or without Bevacizumab in Men with Metastatic Castration-Resistant Prostate Cancer: CALGB 90401. J. Clin. Oncol. 2012, 30, 1534–1540.
- Tannock, I.F.; Fizazi, K.; Ivanov, S.; Karlsson, C.T.; Fléchon, A.; Skoneczna, I.; Orlandi, F.; Gravis, G.; Matveev, V.; Bavbek, S.; et al. Aflibercept versus placebo in combination with docetaxel and prednisone for treatment of men with metastatic castration-resistant prostate cancer (VENICE): A phase 3, double-blind randomised trial. Lancet Oncol. 2013, 14, 760–768.
- Michaelson, M.D.; Oudard, S.; Ou, Y.-C.; Sengeløv, L.; Saad, F.; Houede, N.; Ostler, P.; Stenzl, A.; Daugaard, G.; Jones, R.; et al. Randomized, Placebo-Controlled, Phase III Trial of Sunitinib Plus Prednisone Versus Prednisone Alone in Progressive, Metastatic, Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2014, 32, 76–82.
- Kim, J.W.; McKay, R.R.; Radke, M.R.; Zhao, S.; Taplin, M.-E.; Davis, N.B.; Monk, P.; Appleman, L.J.; Lara, P.N.; Vaishampayan, U.N.; et al. Randomized Trial of Olaparib With or Without Cediranib for Metastatic Castration-Resistant Prostate Cancer: The Results From National Cancer Institute 9984. J. Clin. Oncol. 2023, 41, 871–880.
- Small, E.J.; Tchekmedyian, N.S.; Rini, B.I.; Fong, L.; Lowy, I.; Allison, J.P. A Pilot Trial of CTLA-4 Blockade with Human Anti-CTLA-4 in Patients with Hormone-Refractory Prostate Cancer. Clin. Cancer Res. 2007, 13, 1810–1815.
- McNeel, D.G.; Smith, H.A.; Eickhoff, J.C.; Lang, J.M.; Staab, M.J.; Wilding, G.; Liu, G. Phase I trial of tremelimumab in combination with short-term androgen deprivation in patients with PSA-recurrent prostate cancer. Cancer Immunol. Immunother. 2011, 61, 1137–1147.
- Slovin, S.; Higano, C.; Hamid, O.; Tejwani, S.; Harzstark, A.; Alumkal, J.; Scher, H.; Chin, K.; Gagnier, P.; McHenry, M.; et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: Results from an open-label, multicenter phase I/II study. Ann. Oncol. 2013, 24, 1813–1821.
- Caruso, C. Anti–PD-1–CTLA4 Combo Hits Prostate Cancer. Cancer Discov. 2019, 9, 569–570.
- Beer, T.M.; Kwon, E.D.; Drake, C.G.; Fizazi, K.; Logothetis, C.; Gravis, G.; Ganju, V.; Polikoff, J.; Saad, F.; Humanski, P.; et al. Randomized, Double-Blind, Phase III Trial of Ipilimumab Versus Placebo in Asymptomatic or Minimally Symptomatic Patients With Metastatic Chemotherapy-Naive Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2017, 35, 40–47.
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; Van den Eertwegh, A.J.M.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712.
- Sweeney, C.J.; Gillessen, S.; Rathkopf, D.; Matsubara, N.; Drake, C.; Fizazi, K.; Piulats, J.M.; Wysocki, P.J.; Buchschacher, G.L.; Doss, J.; et al. Abstract CT014: IMbassador250: A phase III trial comparing atezolizumab with enzalutamide vs enzalutamide alone in patients with metastatic castration-resistant prostate cancer (mCRPC). Cancer Res. 2020, 80, CT014. Available online: http://cancerres.aacrjournals.org/content/80/16_Supplement/CT014.abstract (accessed on 18 May 2023).
- Gao, J.; Ward, J.F.; Pettaway, A.C.; Shi, L.Z.; Subudhi, S.K.; Vence, L.M.; Zhao, H.; Chen, J.; Chen, H.; Efstathiou, E.; et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017, 23, 551–555.
- Sharma, P.; Pachynski, R.K.; Narayan, V.; Fléchon, A.; Gravis, G.; Galsky, M.D.; Mahammedi, H.; Patnaik, A.; Subudhi, S.K.; Ciprotti, M.; et al. Nivolumab Plus Ipilimumab for Metastatic Castration-Resistant Prostate Cancer: Preliminary Analysis of Patients in the CheckMate 650 Trial. Cancer Cell 2020, 38, 489–499.e3.
- Gamat-Huber, M.; McNeel, D.G. Androgen deprivation and immunotherapy for the treatment of prostate cancer. Endocr.-Relat. Cancer 2017, 24, T297–T310.
- Stultz, J.; Fong, L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 697–717.
- Di Carlo, E.; D’Antuono, T.; Pompa, P.; Giuliani, R.; Rosini, S.; Stuppia, L.; Musiani, P.; Sorrentino, C. The Lack of Epithelial Interleukin-7 and BAFF/BLyS Gene Expression in Prostate Cancer as a Possible Mechanism of Tumor Escape from Immunosurveillance. Clin. Cancer Res. 2009, 15, 2979–2987.
- Sorrentino, C.; Yin, Z.; Ciummo, S.; Lanuti, P.; Lu, L.-F.; Marchisio, M.; Bellone, M.; Di Carlo, E. Targeting Interleukin(IL)-30/IL-27p28 signaling in cancer stem-like cells and host environment synergistically inhibits prostate cancer growth and improves survival. J. Immunother. Cancer 2019, 7, 201.
- Li, X.-F.; Selli, C.; Zhou, H.-L.; Cao, J.; Wu, S.; Ma, R.-Y.; Lu, Y.; Zhang, C.-B.; Xun, B.; Lam, A.D.; et al. Macrophages promote anti-androgen resistance in prostate cancer bone disease. J. Exp. Med. 2023, 220, e20221007.
- Martori, C.; Sanchez-Moral, L.; Paul, T.; Pardo, J.C.; Font, A.; de Porras, V.R.; Sarrias, M.-R. Macrophages as a Therapeutic Target in Metastatic Prostate Cancer: A Way to Overcome Immunotherapy Resistance? Cancers 2022, 14, 440.
- Di Mitri, D.; Mirenda, M.; Vasilevska, J.; Calcinotto, A.; Delaleu, N.; Revandkar, A.; Gil, V.; Boysen, G.; Losa, M.; Mosole, S.; et al. Re-education of Tumor-Associated Macrophages by CXCR2 Blockade Drives Senescence and Tumor Inhibition in Advanced Prostate Cancer. Cell Rep. 2019, 28, 2156–2168.e5.
- Liu, X.; Tang, J.; Peng, L.; Nie, H.; Zhang, Y.; Liu, P. Cancer-associated fibroblasts promote malignant phenotypes of prostate cancer cells via autophagy. Apoptosis 2023, 1–11.
- Di Donato, M.; Zamagni, A.; Galasso, G.; Di Zazzo, E.; Giovannelli, P.; Barone, M.V.; Zanoni, M.; Gunelli, R.; Costantini, M.; Auricchio, F.; et al. The androgen receptor/filamin A complex as a target in prostate cancer microenvironment. Cell Death Dis. 2021, 12, 1–17.
- Shah, K.; Mallik, S.B.; Gupta, P.; Iyer, A. Targeting Tumour-Associated Fibroblasts in Cancers. Front. Oncol. 2022, 12, 2863.
- Wang, W.; Cheng, B.; Yu, Q. Cancer-associated fibroblasts as accomplices to confer therapeutic resistance in cancer. Cancer Drug Resist. 2022, 5, 889–901.
- Bedeschi, M.; Marino, N.; Cavassi, E.; Piccinini, F.; Tesei, A. Cancer-Associated Fibroblast: Role in Prostate Cancer Progression to Metastatic Disease and Therapeutic Resistance. Cells 2023, 12, 802.
- Di Donato, M.; Cernera, G.; Auricchio, F.; Migliaccio, A.; Castoria, G. Cross-talk between androgen receptor and nerve growth factor receptor in prostate cancer cells: Implications for a new therapeutic approach. Cell Death Discov. 2018, 4, 5.
- Di Donato, M.; Cernera, G.; Migliaccio, A.; Castoria, G. Nerve Growth Factor Induces Proliferation and Aggressiveness in Prostate Cancer Cells. Cancers 2019, 11, 784.
- Di Donato, M.; Giovannelli, P.; Migliaccio, A.; Castoria, G. The nerve growth factor-delivered signals in prostate cancer and its associated microenvironment: When the dialogue replaces the monologue. Cell Biosci. 2023, 13, 60.
- Tolkach, Y.; Kristiansen, G. The Heterogeneity of Prostate Cancer: A Practical Approach. Pathobiology 2018, 85, 108–116.
- Yadav, S.S.; Stockert, J.A.; Hackert, V.; Yadav, K.K.; Tewari, A.K. Intratumor heterogeneity in prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 349–360.
- Boyd, L.K.; Mao, X.; Lu, Y.-J. The complexity of prostate cancer: Genomic alterations and heterogeneity. Nat. Rev. Urol. 2012, 9, 652–664.