Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Alexander Yuile.
Gliomas are the most common primary brain tumors. These cancers are universally fatal with limited treatment options. Glioma cells co-opt non-cancerous cells present in normal brain tissue. This manipulation results in a complex network of cell interactions. This interplay is further complicated by variations depending on specific mutations in glioma cells.
- glioma
- glioblastoma
- tumor microenvironment
- immune microenvironment
1. Introduction
Gliomas are the most common primary brain malignancy [1]. Since 2016, the WHO Tumor classification has dichotomized gliomas by the presence or absence of an IDH1/2 mutation [2]. The wildtype IDH1 enzyme and IDH2 enzymes (encoded for by IDH1 and IDH2, respectively) convert isocitrate to α-KG. In the presence of an IDH1/2 mutation, these enzymes produce 2-hyroxyglutarate (D-2-HG) from isocitrate instead [3,4,5,6][3][4][5][6]. D-2-HG causes a cascade of pro-oncogenic events [6,7][6][7]. In the current WHO 2021 classification, IDH1/2-mutant gliomas are further classified into those with a 1p/19q codeletion (oligodendrogliomas grade 2–3) and those without (astrocytomas grade 2–4). Astrocytic gliomas with high-grade features (either histopathologic or molecular) without an IDH1/2 mutation are termed IDH1/2 wildtype glioblastomas [3].
This adoption of molecular classification signifies an understanding that gliomas with different molecular characteristics behave differently. Compared to wildtype glioblastomas, patients with IDH-mutant astrocytomas are younger at diagnosis and have longer survival. In addition, there is a growing appreciation that this divergent clinical behavior is linked to a complex interdependent relationship with the tumor microenvironment (TME) that varies based on molecular phenotype. Compounding this further is the growing appreciation of non-tumoral, non-immune cells in the TME dynamic, including neuronal and glial modulation of the TME [8].
A better understanding of TME interactions and the impact of molecular phenotype is a crucial step to developing further treatments. For example, gliomas are termed immunologically “cold” because of the predominant immunosuppressive interplay of the TME [9]. This has resulted in immunotherapy being ineffective in gliomas. Immunotherapy has been relatively effective in cancers such as melanomas [10[10][11],11], but immunotherapy trials in gliomas have yet to produce positive clinical trial results [12].
2. Tumor Components of the TME
Glioma cells exist in a network with cells more central to the tumor mass being connected by a synaptic network, with invading tumor cells at the periphery being unconnected [13]. The tumor cell components of the TME comprise stem cells like glioma cells and differentiated glioma cells. The latter forms a heterogenous group defined by differing molecular and transcriptomic signatures [14].2.1. Glioma Stem Cells
These precursor cells exist in the perivascular niche, forming a glioma stem cell pool [15,16][15][16]. The cancer stem cell model argues that a self-renewing population of progenitor cells allows for differentiation into heterogeneous cancer cell populations [17]. Glioma stem cells (GSCs) are widely considered to be resistant to therapy and allow for tumor cell repopulation after therapy [18]. In addition, they have been shown to be extremely plastic, providing a variety of glioma clonal populations and differentiating into supportive cells such as vascular endothelial cells [5,17][5][17]. These factors may explain why there is a positive correlation to stem cell population and tumor grade [4,19][4][19].2.2. Mutational Landscape of Glioma Cells
In general, the most common aberrant molecular pathways are the receptor tyrosine kinase (RTK) pathways (which are further divided into the MAPK-pathway and the AKT/mTOR-pathway), the RB-pathway, and the p53-pathway. Alterations in these pathways tend to be mutually exclusive, and glioma cells tend to harbor an alteration in each of these pathways [6,20,21][6][20][21]. This suggests a highly interactive network of molecular alterations. Reflecting this interplay, it has been recognized that there are recurring patterns in the molecular and mutational landscape. For example, abnormalities of copy number variants occur at a much higher frequency than specific mutations, with deletions having a higher prevalence than amplifications [21]. The most common deletions involve the CDKN2A gene of the RB-pathway, PTEN of the AKT/mTOR-pathway, and NF1 of the MAPK-pathway. While the most common amplifications include the RTK receptors EGFR and PDGFRA, MDM2 and MDM4 of the p53-pathway, PIK3CA of the AKT/mTOR-pathway, and CDK4/6 of the RB pathway, the most frequently observed hotspot mutation is the TP53 gene encoding for p53. Other commonly mutated genes include PTEN, NF1, and EGFR (EGFR mutations usually occur with exon 1–8 aberrancy, which is referred to as EGFRvIII) [20]. Further complicating this landscape, the glioma mutational phenotype is heterogenous and highly plastic. For example, patterns of gene mutations tend to only be seen on recurrence, such as NF1 and TP53 co-mutations, Rb1 and PTEN co-deletions, and LTBP4 gene abnormalities [22]. This clonal change bares consideration as it, in turn, can modify the tumor microenvironment. For example, LTBP4 mutations have been shown to upregulate TGF-beta, which has significant anti-inflammatory properties (discussed below). It is also now recognized that mutational switching occurs in key mutational pathways, such as RTK and p53 pathways, where one pathway mutation is replaced by another on recurrence [6,22,23][6][22][23].2.3. Tumor Cell Subtypes
In addition to molecular features such as IDH status, glioma cells can be classified by their transcriptional profile. Consensus clustering has been used to describe four classes using 840 classifying genes (210 genes per class) [6]. These subtypes, based on prior descriptions and their expression signatures, were named proneural, neural, classical, and mesenchymal [6] (Figure 1).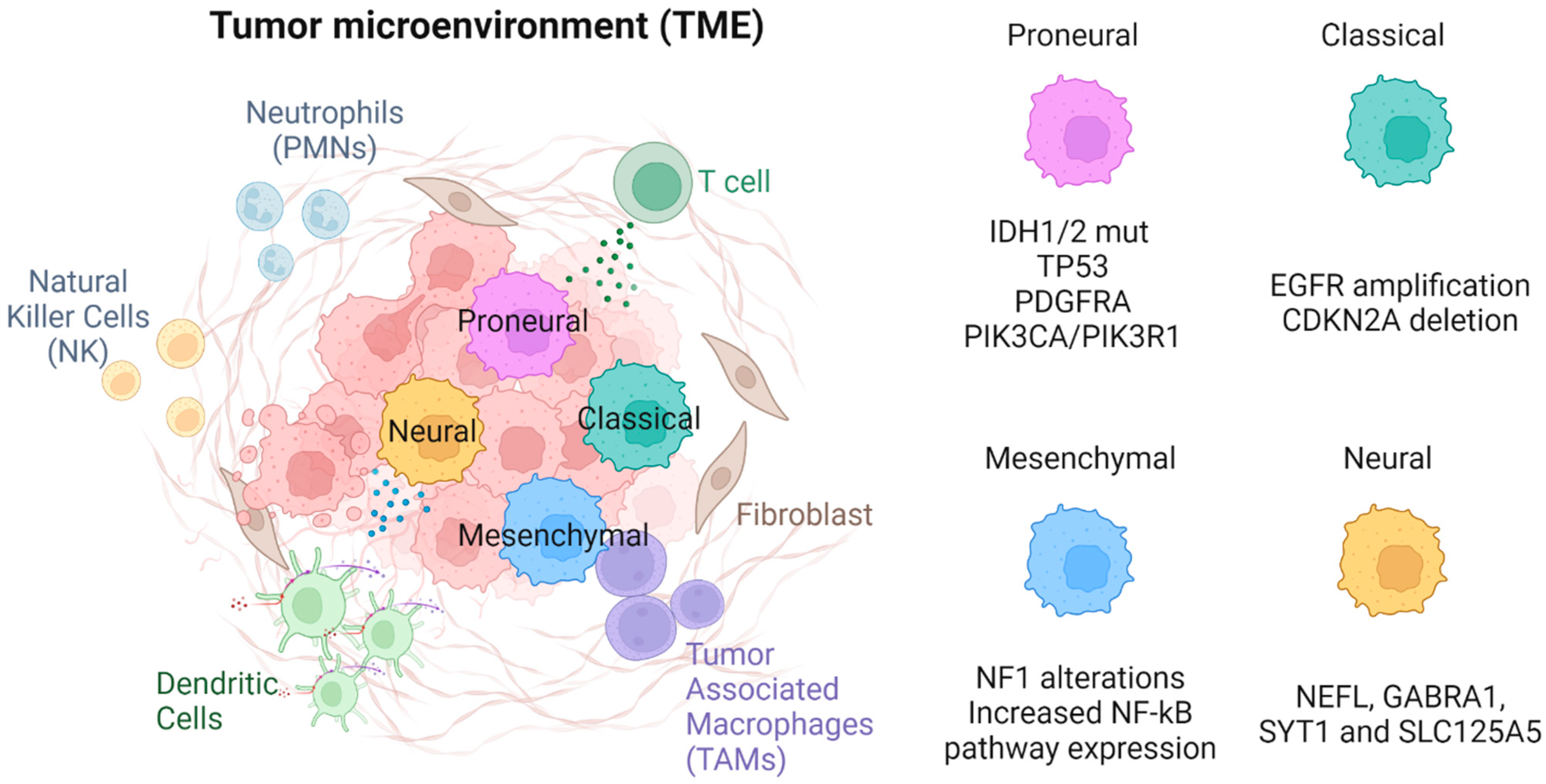
Figure 1. The four GBM molecular subtypes are proneural, neural, classical, and mesenchymal.
2.3.1. Proneural
Proneural subtype gliomas are so named as they have overexpression in multiple proneural development genes such as the SOX genes. They also involve genes associated with development and cell cycle/proliferation [24,25][24][25]. Glioma cells meeting the proneural signature most commonly had IDH1/2 mutations, as well as focal amplification of RTK receptor gene PDGFRA [6,22][6][22]. This PDGFRA amplification is seen almost exclusively in this subtype. Where PDGFRA genes are unaltered, proneural gliomas almost always have increased activity of the genes PIK3CA or PIK3R1 [6]. Unlike classical subtype gliomas, proneural gliomas are also commonly associated with TP53 mutations [6]. Proneural glioma cells have an increased expression of oligodendrocyte development genes, such as the aforementioned PDGFRA, as well as other markers, such as NKX2 and OLIG2 [26]. Adding evidence to the oligodendrocyte phenotype is the expression of SOX10, encoded by a SOX gene, which can induce oligodendrocyte differentiation by antagonizing the NFIA protein. Conversely, NFIA can also inhibit SOX10, leading to astrocyte differentiation. Given SOX genes are overexpressed in the proneural subtype, it is understandable this subtype has an oligodendrocyte-like phenotype [27]. Interestingly, OLIG2 suppresses p21, which is an apoptotic regulator of the p53 pathway [28]. The impact of OLIG2 is an example of how the expression of these oligodendrocyte-associated genes are themselves tumorigenic. Given the strong association between IDH1/2 mutations and the proneural subtype, molecular differences between the proneural subtype compared to other subtypes also serve as a description of the molecular phenotype associated with IDH1/2 mutations.2.3.2. Classical
Glioma cells meeting the “classical” expression pattern were noted to have chromosome 7 amplification and chromosome 10 loss at a high frequency (almost 100% in classical glioblastomas). In addition, most cells had EGFR amplification and homozygous deletion of CDKN2A but were lacking TP53 and IDH mutations and PDGFRA and NF1 alterations when compared to other subtypes [6]. Other mutations commonly seen in classical type gliomas include those of the NOTCH pathway, such as NOTCH3, JAG1, and LFNG, and in the Sonic Hedgehog pathway, such as SMO, GAS1, and GLI2. This subtype is also associated with increased expression of neuronal stem cell markers [6].2.3.3. Mesenchymal
This subtype most notably has NF1 alterations (predominantly hemizygous deletions at 17q11.2), resulting in lower NF1 expression. There is also increased expression of genes of the tumor necrosis superfamily and NF-κB pathways, such as TRADD, RELB, and TNFRSF1A [6,23][6][23]. The increased expression of these genes may explain, in part, why this subtype is the most inflammatory with the highest immune cell burden (discussed below).2.3.4. Neural
The “neural” subtype most commonly has expression of typical neuronal markers such as NEFL, GABRA1, SYT1, and SLC125A5. Genes associated with this subtype are commonly related to neurons, such as those involved in neuronal axial development, neuronal projection, and synaptic transmission [6].3. Immune Components of the TME
A significant proportion of the glioma tumor mass is comprised of immune cells. Our immune system can be generally divided into innate and adaptive arms (Figure 1). The innate immune system provides a rapid response to pathogens but lacks antigenic specificity and immunological memory [7]. The adaptive immune response can mount antigen-specific responses and form memory immune cells [29]. Upon antigenic restimulation of the same antigen, the immune response can increase in speed and magnitude. Both arms are thought to be important in immune surveillance and preventing immune escape by cancerous cells [30]. Although it is long believed that innate and adaptive arms of the immune system are separate entities, there is mounting evidence that there are interplays between the two, generating complex immune responses and forming long-lasting memory cells. Furthermore, immunosuppressive cells play a crucial role in dampening down immune responses to prevent overreaction, especially in the setting of autoimmunity. Immune cells found in the glioma TME include macrophages, neutrophils, dendritic cells (DCs), and natural killer (NK) cells of the innate immune system; CD4+ T cells, CD8+ T cells, and B cells of the adaptive immune system; and immunosuppressive cells such as monocyte-derived suppressor cells (MDSCs) and regulatory T cells (Treg).3.1. Innate Immune Component
3.1.1. Tumor-Associated Macrophages (TAMs)—Microglia and Bone Marrow Derived Macrophages (BMDM)
Approximately 30–40% of the TME are innate immune cells called tumor- associated macrophages (TAMs) [31,32][31][32]. They are formed by two distinct macrophage populations—microglia and BMDM [33,34][33][34]. Microglia are resident macrophage-like cells that are developed from the yolk sac during embryogenesis [35]. They are the only immune cells in the brain at steady state and act as both sentinel immune cells and regulators of homeostasis [36]. Their survival depends on stimulation from colony-stimulation factor (CSF) 1 or interleukin (IL) 34 via its receptor, CSF1R. Although microglia are the only macrophages in a naïve brain, they only compose approximately 15% of the macrophages [37] and reside on the periphery of the TME [38]. The rest of the macrophages found in the TME are BMDM. BMDM do not exist in the brain at steady state. They are circulating monocytes originating from hematopoietic stem cells within our bone marrow or spleen. Upon inflammation, monocytes can migrate to the sites of infection/inflammation and differentiate into macrophages [36,39,40][36][39][40]. Likewise, in gliomas, monocytes can infiltrate via chemotaxis [41[41][42],42], taking advantage of the breakdown of the Blood Brain Barrier (BBB) during glioma pathogenesis [43,44,45,46][43][44][45][46] and through receptor stimulation, differentiating into macrophages. Unlike microglia, BMDM are found intratumorally [38,47][38][47]. Within the TME, BMDM exist on a spectrum of polarization between M1 (pro-inflammatory/immune-stimulatory) and M2 (anti-inflammatory/ immunosuppressive). Glioma cells recruit peripheral monocytes and polarize them toward the M2 phenotype. The degree of BMDM recruitment correlates positively with glioma grade and progression [34] and negatively with prognosis [48].3.1.2. Neutrophils (PMNs)
Although neutrophils are the most abundant leukocytes in our blood stream, they are not a major component within the glioma TME. Under steady state, they are generated from haemopoietic stem cells from our bone marrow. During infection, neutrophils can exert multiple actions, such as phagocytosis, degranulation, release of neutrophil extracellular trap, and antigen presentation, in order to control the spread of foreign invaders [49]. Like BMDM, neutrophils correlate with prognosis negatively, and their role in gliomas is starting to be recognized [50]. A recent study identified a unique population of myeloperoxidase (MPO)-positive macrophages associated with long-term survival [51]. Neutrophils express abundant MPO, and one could infer that these macrophages could be engulfing neutrophils, but this may reflect a more complex process.3.1.3. Dendritic Cells
Dendritic cells (DCs) are a diverse group of myelocytes that are well known for their ability to survey nearby environments, uptake and process antigens, and activate T cells. For this, they are known as one of the professional antigen-presenting cells (APCs), linking our innate and adaptive immune systems, and they exist within the glioma TME [52,53][52][53]. Their role in the TME is not yet elucidated, but there seems to be a positive correlation between the frequency of TILs with DCs within the TME [52].3.1.4. Natural Killer (NK) Cells
NK cells are innate cells with cytotoxic capabilities, and their presence in the glioma TME has been described [54]. Unlike cytotoxic T cells (discussed later), NK cells detect targets for killing by a mechanism called “missing self” instead of antigenic stimulation. Foreign invaders, such as bacteria or viruses, can downregulate MHC class I molecules and avoid cytotoxic T cell recognition. NK cells can detect the downregulation of MHC class I molecules and are actioned to kill. Tumor cells can downregulate MHC class I molecules as a mechanism of immune escape, but NK cells can potentially kill these tumor cells. It has been shown that chemokines secreted by glioma cells can attract NK cell infiltration to the TME, and this is associated with better prognosis [55].3.1.5. Monocyte-Derived Suppressor Cells (MDSCs)
MDSCs are a heterogenous group of cells with their main function of putting a break on our immune system. There are two main types of MDSCs—monocytic and polymorphonuclear (PMN). Both types are found in the glioma TME [56], and their numbers correlate negatively with prognosis [57].3.2. Adaptive Immune Component
Tumor-Infiltrating lymphocytes (TILs)
TILs found in the TME consist of NK (described above), CD4+ T, CD8+ T, and B cells. CD4+ T cells, also known as helper T cells, orchestrate our adaptive immune system. They are activated by professional APCs and differentiate into T helper 1 (Th1), T helper 2 (Th2), and T helper 17 (Th17) cells depending on the stimulating cytokines. Th1 cells can polarize the environment towards cellular immunity (CD8+ T cell response). When activated, CD8+ T cells cause cellular damage to target cells. Thus, they are also known as cytotoxic T cells. They kill cells via multiple mechanisms—cytokine (IFN-g and TNF-a) secretion, FAS-ligand-receptor signaling, and perforin and granzyme release [58]. Th2 cells can polarize the environment towards B cell-mediated humoral immunity (antibody response). Th17 cells are usually associated with autoimmunity but may also play an anti-tumoral role [59]. Unlike the abundance of TAMs in the glioma TME, TILs are scarce and comprise only 0.25% of the cells. T cells seem to home to bone marrow rather than the TME [60]. Of these, the majority are functionally exhausted and ineffective [61,62][61][62]. Furthermore, anti-tumor lymphocytes are drastically reduced (25% of the already depleted lymphocyte population) [63]. This contrasts heavily with tumors with a favorable immunotherapy response which have a comparatively higher number of lymphocyte infiltration. Regulatory T cells are also TILs found in the glioma TME. Unlike the other lymphocytes, regulatory T cells are immunosuppressive and confer a poor prognosis in glioma patients.References
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2021, 23 (Suppl. S3), iii1–iii105.
- Pekmezci, M.; Rice, T.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Hansen, H.; Sicotte, H.; Kollmeyer, T.M.; McCoy, L.S.; Sarkar, G.; et al. Adult infiltrating gliomas with WHO 2016 integrated diagnosis: Additional prognostic roles of ATRX and TERT. Acta Neuropathol. 2017, 133, 1001–1016.
- WHO. Classification of Tumours Editorial Board. Central Nervous System Tumours. 2021. Available online: https://publications.iarc.fr/Book-And-Report-Series/Who-Classification-Of-Tumours/Central-Nervous-System-Tumours-2021 (accessed on 27 January 2023).
- Bradshaw, A.; Wickremsekera, A.; Tan, S.T.; Peng, L.; Davis, P.F.; Itinteang, T. Cancer Stem Cell Hierarchy in Glioblastoma Multiforme. Front. Surg. 2016, 3, 21.
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828.
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. An integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 2010, 17, 98.
- Medzhitov, R.; Janeway, C.A. Innate immunity: The virtues of a nonclonal system of recognition. Cell 1997, 91, 295–298.
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495.
- Desland, F.A.; Hormigo, A. The CNS and the Brain Tumor Microenvironment: Implications for Glioblastoma Immunotherapy. Int. J. Mol. Sci. 2020, 21, 7358.
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492.
- Long, G.V.; Atkinson, V.; Lo, S.; Sandhu, S.; Guminski, A.D.; Brown, M.P.; Wilmott, J.S.; Edwards, J.; Gonzalez, M.; Scolyer, R.A.; et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: A multicentre randomised phase 2 study. Lancet Oncol. 2018, 19, 672–681.
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs. Bevacizumab in Patients With Recurrent Glioblastoma. JAMA Oncol. 2020, 6, 1003–1010.
- Venkataramani, V.; Yang, Y.; Schubert, M.C.; Reyhan, E.; Tetzlaff, S.K.; Wißmann, N.; Botz, M.; Soyka, S.J.; Beretta, C.A.; Pramatarov, R.L.; et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 2022, 185, 2899–2917.
- Becker, A.P.; Sells, B.E.; Haque, S.J.; Chakravarti, A. Tumor Heterogeneity in Glioblastomas: From Light Microscopy to Molecular Pathology. Cancers 2021, 13, 761.
- Charles, N.; Holland, E.C. The perivascular niche microenvironment in brain tumor progression. Cell Cycle 2010, 9, 3012–3021.
- Pietras, A.; Katz, A.M.; Ekström, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014, 14, 357–369.
- Piper, K.; DePledge, L.; Karsy, M.; Cobbs, C. Glioma Stem Cells as Immunotherapeutic Targets: Advancements and Challenges. Front. Oncol. 2021, 11, 615704.
- Gallego-Perez, D.; Chang, L.; Shi, J.; Ma, J.; Kim, S.-H.; Zhao, X.; Malkoc, V.; Wang, X.; Minata, M.; Kwak, K.J.; et al. On-Chip Clonal Analysis of Glioma-Stem-Cell Motility and Therapy Resistance. Nano Lett. 2016, 16, 5326–5332.
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82.
- Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068.
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477.
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.S.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.-J.; et al. Clonal Evolution of Glioblastoma under Therapy. Nat. Genet 2016, 48, 768–776.
- Di Cintio, F.; Dal Bo, M.; Baboci, L.; De Mattia, E.; Polano, M.; Toffoli, G. The Molecular and Microenvironmental Landscape of Glioblastomas: Implications for the Novel Treatment Choices. Front. Neurosci. 2020, 14, 603647.
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173.
- Whitfield, M.L.; Sherlock, G.; Saldanha, A.J.; Murray, J.I.; Ball, C.A.; Alexander, K.E.; Matese, J.C.; Perou, C.M.; Hurt, M.M.; Brown, P.O.; et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell 2002, 13, 1977–2000.
- Noble, M.; Pröschel, C.; Mayer-Pröschel, M. Getting a GR(i)P on oligodendrocyte development. Dev. Biol. 2004, 265, 33–52.
- Glasgow, S.M.; Zhu, W.; Stolt, C.C.; Huang, T.-W.; Chen, F.; LoTurco, J.J.; Neul, J.L.; Wegner, M.; Mohila, C.; Deneen, B. Mutual antagonism between Sox10 and NFIA regulates diversification of glial lineages and glioma subtypes. Nat. Neurosci. 2014, 17, 1322–1329.
- Ligon, K.L.; Huillard, E.; Mehta, S.; Kesari, S.; Liu, H.; Alberta, J.A.; Bachoo, R.M.; Kane, M.; Louis, D.N.; Depinho, R.A.; et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 2007, 53, 503–517.
- Burnet, F.M. The Clonal Selection Theory of Acquired Immunity; Vanderbilt University Press: Nashville, TN, USA, 1959; p. 232. Available online: https://www.biodiversitylibrary.org/item/34425 (accessed on 17 December 2022).
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570.
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2012, 60, 502–514.
- Engler, J.R.; Robinson, A.E.; Smirnov, I.; Hodgson, J.G.; Berger, M.S.; Gupta, N.; James, C.D.; Molinaro, A.; Phillips, J.J. Increased Microglia/Macrophage Gene Expression in a Subset of Adult and Pediatric Astrocytomas. PLoS ONE 2012, 7, e43339.
- Bowman, R.L.; Joyce, J.A. Therapeutic targeting of tumor-associated macrophages and microglia in glioblastoma. Immunotherapy 2014, 6, 663–666.
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27.
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551.
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468.
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278.
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234.
- Mysore, V.; Tahir, S.; Furuhashi, K.; Arora, J.; Rosetti, F.; Cullere, X.; Yazbeck, P.; Sekulic, M.; Lemieux, M.E.; Raychaudhuri, S.; et al. Monocytes transition to macrophages within the inflamed vasculature via monocyte CCR2 and endothelial TNFR2. J. Exp. Med. 2022, 219, e20210562.
- Trouplin, V.; Boucherit, N.; Gorvel, L.; Conti, F.; Mottola, G.; Ghigo, E. Bone marrow-derived macrophage production. J. Vis. Exp. 2013, 81, e50966.
- Jung, Y.; Ahn, S.-H.; Park, H.; Park, S.H.; Choi, K.; Choi, C.; Kang, J.L.; Choi, Y.-H. MCP-1 and MIP-3α Secreted from Necrotic Cell-Treated Glioblastoma Cells Promote Migration/Infiltration of Microglia. Cell Physiol. Biochem. 2018, 48, 1332–1346.
- Liu, X.; Liu, Y.; Qi, Y.; Huang, Y.; Hu, F.; Dong, F.; Shu, K.; Lei, T. Signal Pathways Involved in the Interaction Between Tumor-Associated Macrophages/TAMs and Glioblastoma Cells. Front. Oncol. 2022, 12, 822085.
- Chen, Z.; Ross, J.L.; Hambardzumyan, D. Intravital 2-photon imaging reveals distinct morphology and infiltrative properties of glioblastoma-associated macrophages. Proc. Natl. Acad. Sci. USA 2019, 116, 14254–14259.
- Feng, S.; Cen, J.; Huang, Y.; Shen, H.; Yao, L.; Wang, Y.; Chen, Z. Matrix metalloproteinase-2 and -9 secreted by leukemic cells increase the permeability of blood-brain barrier by disrupting tight junction proteins. PLoS ONE 2011, 6, e20599.
- Ishihara, H.; Kubota, H.; Lindberg, R.L.P.; Leppert, D.; Gloor, S.M.; Errede, M.; Virgintino, D.; Fontana, A.; Yonekawa, Y.; Frei, K. Endothelial cell barrier impairment induced by glioblastomas and transforming growth factor beta2 involves matrix metalloproteinases and tight junction proteins. J. Neuropathol. Exp. Neurol. 2008, 67, 435–448.
- Schneider, S.W.; Ludwig, T.; Tatenhorst, L.; Braune, S.; Oberleithner, H.; Senner, V.; Paulus, W. Glioblastoma cells release factors that disrupt blood-brain barrier features. Acta Neuropathol. 2004, 107, 272–276.
- Pinton, L.; Masetto, E.; Vettore, M.; Solito, S.; Magri, S.; D’Andolfi, M.; Del Bianco, P.; Lollo, G.; Benoit, J.-P.; Okada, H.; et al. The immune suppressive microenvironment of human gliomas depends on the accumulation of bone marrow-derived macrophages in the center of the lesion. J. Immunother. Cancer 2019, 7, 58.
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774.
- Li, Y.; Wang, W.; Yang, F.; Xu, Y.; Feng, C.; Zhao, Y. The regulatory roles of neutrophils in adaptive immunity. Cell Commun. Signal. 2019, 17, 147.
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; de Groot, J.F. Neutrophils Promote the Malignant Glioma Phenotype through S100A4. Clin. Cancer Res. 2014, 20, 187–198.
- Karimi, E.; Yu, M.W.; Maritan, S.M.; Perus, L.J.M.; Rezanejad, M.; Sorin, M.; Dankner, M.; Fallah, P.; Doré, S.; Zuo, D.; et al. Single-cell spatial immune landscapes of primary and metastatic brain tumours. Nature 2023, 614, 555–563.
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17.
- Yang, I.; Han, S.J.; Sughrue, M.E.; Tihan, T.; Parsa, A.T. Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: Evidence of distinct immunological microenvironments that reflect tumor biology. J. Neurosurg. 2011, 115, 505–511.
- Ren, F.; Zhao, Q.; Huang, L.; Zheng, Y.; Li, L.; He, Q.; Zhang, C.; Li, F.; Maimela, N.R.; Sun, Z.; et al. The R132H mutation in IDH1 promotes the recruitment of NK cells through CX3CL1/CX3CR1 chemotaxis and is correlated with a better prognosis in gliomas. Immunol. Cell Biol. 2019, 97, 457–469.
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Bossman, S.A.J.F.H.; Ter Laan, M.; Wesseling, P.; Adema, G.J. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro Oncol. 2016, 18, 1253–1264.
- Richard, S.A. Explicating the Pivotal Pathogenic, Diagnostic, and Therapeutic Biomarker Potentials of Myeloid-Derived Suppressor Cells in Glioblastoma. Dis. Markers 2020, 2020, 8844313.
- Halle, S.; Halle, O.; Förster, R. Mechanisms and Dynamics of T Cell-Mediated Cytotoxicity In Vivo. Trends Immunol. 2017, 38, 432–443.
- Paladugu, M.; Thakur, A.; Lum, L.G.; Mittal, S.; Parajuli, P. Generation and immunologic functions of Th17 cells in malignant gliomas. Cancer Immunol. Immunother. 2013, 62, 75–86.
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468.
- Fecci, P.E.; Sweeney, A.E.; Grossi, P.M.; Nair, S.K.; Learn, C.A.; Mitchell, D.A.; Cui, X.; Cummings, T.J.; Bigner, D.D.; Gilboa, E.; et al. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin. Cancer Res. 2006, 12 Pt 1, 4294–4305.
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin. Cancer Res. 2018, 24, 3792–3802.
- Han, S.; Ma, E.; Wang, X.; Yu, C.; Dong, T.; Zhan, W.; Wei, X.; Liang, G.; Feng, S. Rescuing defective tumor-infiltrating T-cell proliferation in glioblastoma patients. Oncol. Lett. 2016, 12, 2924–2929.
More