The entry summarizes the roles of Nurr1 in a wide range of tumors and the underlying pathways for carcinogenesis.
The entry summarizes the roles of Nurr1 in a wide range of tumors and the underlying pathways for carcinogenesis.
Nuclear receptor related-1 protein (Nurr1), coded by an early response gene, is involved in multiple cellular and physiological functions, including proliferation, survival, and self-renewal. Dysregulation of Nurr1 has been frequently observed in many cancers and is attributed to multiple transcriptional and post-transcriptional mechanisms.
- Nurr1
- NR4A2
- signaling pathway
- carcinogenesis
- cancer biology
- tumor immunology
- immunosuppression
1. Introduction
Nuclear receptor related-1 protein (Nurr1), also known as NR4A2, belongs codedto the nuclear receptor (NR) subfamily 4 group A (NR4A). Nurr1 is classified as an ‘orphan’ NR due to the absence of known natural ligands[1]. byThese NRs an re considered as early response gene, is invs that respond to many different signals, including fatty acids and hormones [2]. Crosstalved in multiplek between Nurr1 and various signaling pathways further modulates various cellular and physiological functionresponses, including proliferation, survivalinvasion, and self-renewalapoptosis. Dysregulation of Nurr1 has been is frequently observed in many different types of cancers and is attributed to multiple transcriptional and post-transcriptional mechanisms. Besides, Nurr1 exhibi, with studies demonstrating either pro-oncogenic or tumor-suppressor roles in different contexts [2][3]. Evidextensive crosstalk with many oncogenic and tumorce suggesting that suppressor molecules, which contribute to its potential pro-malignant behaviors. Furthermore,ion of Nurr1 maintains the pluripotency of hematopoietic stem cells supports the contention that the Nurr1 is a key player in attenuating antitumor immune responses. (draft for definition)family is important in maintaining terminal differentiation of epithelia [4].
Introduction
Nuclear receptor related-1 protein (Nurr1), also known as NR4A2, belongs to the nuclear receptor (NR) subfamily 4 group A (NR4A). Nurr1 is classified as an ‘orphan’ NR due to the absence of known natural ligands [1]. These NRs are considered as early response genes that respond to many different signals, including fatty acids and hormones [2]. Crosstalk between Nurr1 and various signaling pathways further modulates various cellular and physiological responses, including proliferation, invasion, and apoptosis. Dysregulation of Nurr1 is frequently observed in different types of cancer, with studies demonstrating either pro-oncogenic or tumor-suppressor roles in different contexts [2,3]. Evidence suggesting that suppression of Nurr1 maintains the pluripotency of hematopoietic stem cells supports the contention that the Nurr1 family is important in maintaining terminal differentiation of epithelia [4].
Members of the NR4A family (Nurr1, Nur77, NOR-I) are well-conserved in their genomic organization with ≈91–95% similarity in their DNA binding domain (DBD) and ≈60% similarity in the C-terminal ligand-binding domain (LBD) [2,5][2][5]. The N-terminal domain of NR4A members contains an activation function-1 (AF-1), which mediates ligand-independent transactivation of the NR [6][6]. The DBD is responsible for recognizing the response element and binding to the promoter. Besides, the C-terminal domain is composed of LBD and the ligand-dependent activation function-2 (AF-2) (Figure 1A). The LBD and its amino acid composition define the interaction of the NR with the ligand. Unlike conventional NRs, which have a hydrophobic cleft in the LBD for binding of a coactivator or corepressor, the Nurr1 LBD is tightly packed with hydrophobic side chains, leading to the absence of or atypical ligand-binding cavity clefts [7][7]. Although the mechanism of regulation of Nurr1-mediated transcription remains unknown, mounting evidence suggests that post-translational and protein-protein interactions are crucial in transcriptional regulation and that the AF-1 domain is involved in ligand recruitment [2,3][2][3].
Nurr1 is considered a constitutively active transcription factor due to the constitutive and cell-type-specific activity of its LBD [7][7]. Nurr1 induces gene expression by binding as a monomer to the nerve growth factor-induced clone B (NGFI-B) response element (NBRE) with common octanucleotide binding site 5¢-AAAGGTCA-3¢[2] [2] and as homodimers or heterodimers with Nur77 to the Nur response element (NurRE; 5′-TGACCTTT-n6-AAAGGTCA-3′) [6,8] [6][8]. Nurr1 also heterodimerizes with retinoid X receptor (RXR) and binds to the direct repeat element with five spacer nucleotide element (DR5; 5¢-GGTTCA-n5-AGGTCA-3¢) to regulate cell proliferation and survival; this suggests crosstalk of Nurr1 with retinoic acid signaling[9] [9] (Figure 1B). An extensive review of the structure of Nurr1 is summarized by Maxwell et al. [2][2].
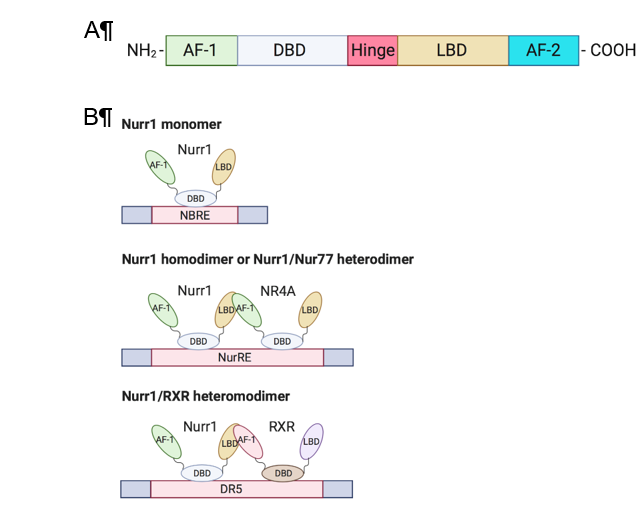
Figure 1. Structure and DNA-binding elements of Nurr1. (A) Nurr1 structure (B) DNA-binding elements of monomeric, homodimeric, and heterodimeric Nurr1. AF: activation function; DBD: DNA-binding domain; LBD: ligand-binding domain; NRBE: nerve growth factor-induced clone B (NGFI-B) response element; NurRE: Nur response element; DR5: direct repeat element with five spacer nucleotides element; RXR: retinoid X receptor.
Expression and Function of Nurr1 in Cancer
2. Expression and Function of Nurr1 in Cancer
Nurr1 promotes or suppresses cancer progression depending on the cellular context, and its oncogenic roles have been reported in many cancers. Overexpression of Nurr1 has been shown to promote cancer cell proliferation, invasion and anchorage-independent growth [10,11]10][11]. It also enhances cell survival by suppressing apoptosis [12,13]12][13]. Apart from augmenting cancer aggressiveness, Nurr1 confers therapeutic resistance to cancer cells, including radioresistance and chemoresistance to 5-fluorouracil [14–16][14][15][16]. Several pro-inflammatory molecules including prostaglandin E2 and thromboxane A2 were reported to induce Nurr1 expression, which can lead to carcinogenesis [17,18][17][18]. Furthermore, the expression of Nurr1 is correlated with different clinicopathological parameters in multiple cancers [12–14]12][13][14]. Since Nurr1 is generally considered to be a transcription factor, its roles in the cytosol are not well characterized. Immunohistochemical staining of Nurr1 revealed its localization in cytosol, and that the level of cytoplasmic Nurr1 correlates with survival and tumor grade in bladder and cervical cancer, respectively [16,19][16][19]. In contrast, tumor-suppressive roles of Nurr1 have been described in gastric cancer [20][20]. Since functions of Nurr1 are cell type-dependent, the roles of Nurr1 in tumorigenesis are summarized and discussed according to specific cancer types.
2.1. Breast Cancer
Early studies on the role of Nurr1 in cancer began in 1997 when Maruyama et al. first showed that the NR4A family is closely related to retinoic acid signaling in the breast cancer cell line MCF-7 [21][21]. Although not many studies on the role of Nurr1 in cancer have been conducted since then, the association of Nurr1 with cancer is supported by some recent work. In 2013, Llpois et al. showed that Nurr1 has a dichotomous role in breast cancer [22][22]. Stronger Nurr1 staining was observed in normal breast epithelia compared with breast carcinoma cells, with no correlation with grade or stage, but was positively correlated with relapse-free survival. Moreover, there is an inverse correlation between the expression of Nurr1 and p53 in primary cancer. Paradoxically, silencing of Nurr1 attenuates breast tumor xenograft growth and metastasis in vivo. Taken together with its high expression in normal epithelia, Nurr1 may play a role in maintaining a differentiated epithelial phenotype in normal, non-proliferating breast epithelium, yet acquire tumorigenic capability in transformed tissues [22][22]. Table 1 gives a brief overview of Nurr1 expression and its associated functions and clinicopathological correlation in different cancers.
Table 1. Summary of Nurr1 expression and its associated functions and clinicopathological correlation in cancers.
|
Cancer |
Nurr1 Expression |
Functions |
Stage Correlation |
Survival Correlation |
Ref. |
|
Breast |
Low |
Inhibited tumor growth and metastasis in vivo * |
No ^ |
Yes^ |
[22] |
|
Bladder |
High |
Promoted migration and tumor growth in vivo |
Yes ^^^ |
Yes^^^ |
|
|
Colon |
High |
Promoted cell proliferation, migration, and chemoresistance to 5-fluorouracil |
No ^^ |
Yes^^ |
|
|
Gastric |
Low |
Promoted apoptosis and inhibited gastrin-induced migration and invasion |
NA |
NA |
|
|
High |
Promoted tumor growth in vivo and chemoresistance to 5-fluorouracil |
No ^^ |
Yes ^^ |
[14] |
|
|
Cervical |
High |
Promoted anchorage-independent growth, anoikis |
NA |
NA |
|
|
Prostate |
High |
Promoted cell proliferation, migration, invasion, and resistance to apoptosis |
Yes ^ |
NA |
[12] |
|
Pancreatic |
High |
Promoted cell proliferation and resistance to apoptosis |
Yes ^ |
Yes ^ |
[13] |
|
Brain |
High |
Promoted cell proliferation, migration, invasion and survival |
NA |
Yes # |
[26] |
|
Cancer |
Nurr1 Expression |
Functions |
Stage Correlation |
Survival Correlation |
Ref. |
|
Breast |
Low |
Inhibited tumor growth and metastasis in vivo * |
No ^ |
Yes^ |
[22] |
|
Bladder |
High |
Promoted migration and tumor growth in vivo |
Yes ^^^ |
Yes^^^ |
[19,23] |
|
Colon |
High |
Promoted cell proliferation, migration, and chemoresistance to 5-fluorouracil |
No ^^ |
Yes^^ |
[10,15] |
|
Gastric |
Low |
Promoted apoptosis and inhibited gastrin-induced migration and invasion |
NA |
NA |
[20,24] |
|
High |
Promoted tumor growth in vivo and chemoresistance to 5-fluorouracil |
No ^^ |
Yes ^^ |
[14] |
|
|
Cervical |
High |
Promoted anchorage-independent growth, anoikis |
NA |
NA |
[11,25] |
|
Prostate |
High |
Promoted cell proliferation, migration, invasion, and resistance to apoptosis |
Yes ^ |
NA |
[12] |
|
Pancreatic |
High |
Promoted cell proliferation and resistance to apoptosis |
Yes ^ |
Yes ^ |
[13] |
|
Brain |
High |
Promoted cell proliferation, migration, invasion and survival |
NA |
Yes # |
[26] |
* Dichotomous role of Nurr1 was reported: High expression of Nurr1 in normal breast cancer in immunohistochemistry, yet silencing of Nurr1 inhibited tumor growth and metastasis in nude mice; ^ Analyzed with total Nurr1; ^^ Analyzed with nuclear Nurr1; ^^^ Analyzed with cytoplasmic Nurr1; # Analyzed with Nurr1 mRNA; NA: data not available; Ref.: reference.
2.2. Bladder Cancer
Nurr1 functions as a transcription factor and localizes in the nucleus. Studies on the localization of Nurr1 and its roles other than that of a transcription factor are sparse. The problem associated with Nurr1 cytoplasmic mislocalization has been addressed by Inamoto et al. [19]. Total Nurr1 expression is shown to positively correlate with stage, grade, and metastasis of bladder cancer; Nurr1 staining in the cytosol is widely observed in primary bladder cancer and barely detected in the normal epithelium. Since Nurr1 is presumed to reside in the nucleus, expression of cytoplasmic Nurr1 is found to correlate with decreased disease-specific survival and recurrence-free survival in patients. Such correlation is not observed when total Nurr1 or nuclear Nurr1 staining is used for analysis. Furthermore, silencing of Nurr1 attenuates the migration of cancer cells. Although the reason for the oncogenic phenotype of Nurr1 in the cytoplasm remains poorly understood, Nurr1 may use mechanisms similar to its homolog Nur77 to exert opposing biological activities. This was demonstrated by Lin et al., who found that Nur77 shuttles from the nucleus to the cytoplasm to interact with Bcl-2 and subsequently switches from an anti-apoptotic to a pro-apoptotic function [27][27]. Hence, it was suggested that Nurr1 ceases to act as a transcription factor when it translocates to the cytoplasm.
Nurr1’s role as a potential therapeutic target for bladder cancer was the subject of another study by Inamoto et al., which showed that diindolylmethane (DIM)-C-pPhCl (a new class of methylene-substituted DIM) activates the LBD of Nurr1, thereby attenuating the tumorigenic properties of bladder cancer cells [23]. DIM-C-pPhCl induces DNA fragmentation and causes regression of orthotopic bladder cancer in a tumor xenograft model.
2.3. Gastrointestinal Cancer
Prostaglandin E2 (PGE2) is a pro-inflammatory bioactive lipid produced by colorectal cancer cells that promotes tumor growth by binding to G protein-coupled receptors [17,28][17][28]. Vijaykumar et al. demonstrated that PGE2 induces Nurr1 expression and subsequently blocks apoptosis in colorectal cancer cells [17][17]. Nurr1 was found to be highly expressed in Apc+/− mouse adenomas and sporadic colorectal carcinoma as compared to normal intestinal mucosa. In addition, cyclooxygenase-2 (COX-2), from which PEG2 is derived, is associated with poor prognosis in colorectal cancer and is positively correlated with Nurr1 expression [17,29][17][29]. Nurr1 shares similar regions of localization with Ki67 and is mainly found in the proliferation crypt of mice intestinal tissue.
The role of Nurr1 and COX-2 was further studied by Zagani et al. Treatment with a COX-2 inhibitor, parecoxib, downregulates Nurr1 and osteopontin (OPN), a colon cancer progression marker. OPN expression is associated with poor prognosis, lymphatic metastasis, and higher TNM stage in colorectal cancer [30,31][30][31]. Although Nurr1 expression increases in tumor tissues, unlike OPN, its expression does not correlate with tumor stage in colorectal cancer [10][10].
High expression of vascular endothelial growth factor (VEGF) is crucial to tumor growth and angiogenesis and is frequently reported in gastrointestinal cancers [32][32]. Zhao et al. showed that VEGF induces Nurr1 promoter activity, and mRNA and protein expression, by facilitating binding of cAMP response element binding protein (CREB) to the Nurr1 promoter in endothelial cells [33][33].
To summarize, Nurr1 is closely related to COX-2, OPN, and VEGF. Aberrant expression of all or either one of these proteins may dysregulate the expression of their interacting partners and act synergistically to promote inflammation to promote development of gastrointestinal cancers.
Unlike many other gastrointestinal cancers, Nurr1 is reported to be tumor-suppressive in gastric cancer [20,24][20][24]. Chang et al. reported that Nurr1 is significantly downregulated in primary gastric cancer compared with the normal gastric mucosa; it is also downregulated in synchronous liver metastasis compared with the paired gastric cancer [24]. Another study by Misund et al. demonstrated that endogenous Nurr1 enhances apoptosis and attenuates gastrin-induced invasion [20][20]. Nurr1 is shown to be negatively regulated by two gastrin-induced proteins: (i) inducible cAMP early repressor (ICER), which represses Nurr1 transcription; and (ii) zinc finger protein 36, C3H1 type-like 1 (Zfp36l1), which degrades and reduces Nurr1 mRNA levels. Silencing of Nurr1 promotes cancer cell migration and the effect is further augmented in response to gastrin treatment. Similar to what is observed in bladder cancer, as discussed above, Nurr1 cytoplasmic localization is observed in gastric cancer by gastrin-mediated nucleus-cytosol shuttling, but the role of cytoplasmic Nurr1 remains to be elucidated. In contrast, Han et al. demonstrated that ectopic overexpression of Nurr1 enhances gastric cancer formation in vivo [14][14]. It also confers chemoresistance to 5-fluorouracil by attenuating 5-fluorouracil-induced apoptosis. High Nurr1 expression is associated with unfavorable prognosis, particularly in those receiving chemotherapy. Therefore, the role of Nurr1 in gastric cancer remains controversial and more functional characterization is needed to understand its contribution to gastric carcinogenesis.
Signaling Pathways Regulating Nurr1 Expression
3. Signaling Pathways Regulating Nurr1 Expression
Aberrant signaling pathways are observed in the transformation and maintenance of malignant phenotypes of tumors. Since Nurr1 is frequently associated with either oncogenic or tumor-suppressive properties in different contexts, it is logical to propose that dysregulation of signaling pathways affects Nurr1 expression and promotes cancer development. The promoter of Nurr1 contains a highly conserved sequence of cAMP response element (CRE) and kappa B (κB) site. Mounting evidence indicates that multiple signaling events eventually converge to promote the phosphorylation of CRE binding protein (CREB) and nuclear factor kappa B (NF-κB) to enhance the transcriptional activity of Nurr1 [39][19]. Although transcriptional regulation is considered as the primary method to regulate Nurr1 expression, post-transcriptional regulation by microRNA (miR) also exerts an impact on the translation of Nurr1 mRNA [40][37].
The interplay of Nurr1 with the TXA2 pathway, PGE2 pathway, VEGF, and miR at the transcriptional and posttranscriptional levels will be summarized in the following section. Figure 2 and Table 2 summarize the interplay of Nurr1 with different signaling pathways in cancers.
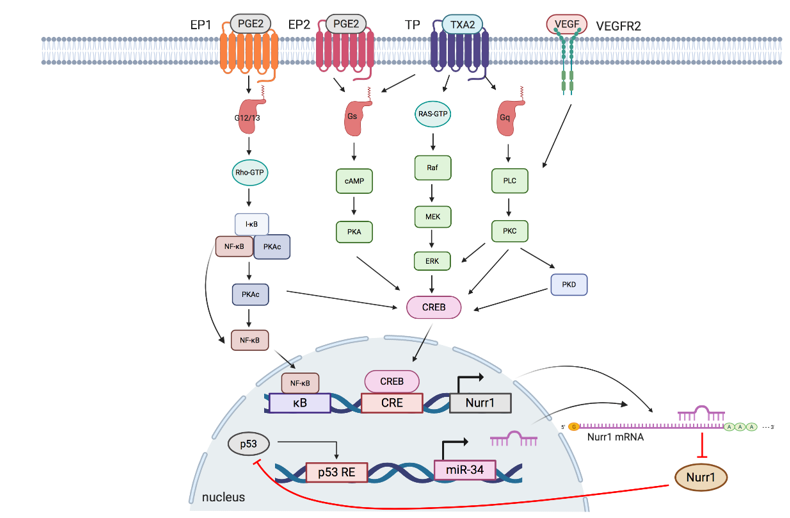
Figure 2. Schematic overview of signaling pathways regulating Nurr1 expression. TXA2, PGE2, and VEGF pathways modulate phosphorylation of NF-κB and CREB to regulate the transcriptional activity of Nurr1. p53/microRNA-34/Nurr1 forms a positive feedback loop to suppress p53 activation and the subsequent expression of microRNA-34, leading to the upregulation of Nurr1 protein expression. EP: prostaglandin E2 receptor; PGE2: prostaglandin E2; TP: thromboxane A2 receptors: TP; TXA2: thromboxane A2: VEGFR: vascular endothelial growth factor receptor; VEGF: vascular endothelial growth factor; G12/13: G12/G13 alpha subunit; Gs: Gs alpha subunit; Gq: Gq alpha subunit; κB: kappa-B; CRE: cAMP response elements; p53 RE: cAMP p53 response element.
Table 2. Signaling pathways regulating Nurr1 expression in cancer development.
|
Pathway |
Cancer |
Mode of Regulation |
|
TXA2 pathway |
Lung cancer [18] |
Transcriptional regulation |
|
PGE2 pathway |
Neuroblastoma[38]; Colorectal cancer[17]; Lung cancer[39] |
Transcriptional regulation |
|
p53/miR-34/Nurr1 loop |
Colorectal cancer[38] |
Post-transcriptional regulation |
|
VEGF/PKD pathway |
Endothelia angiogenesis [33] |
Transcriptional regulation |
|
Pathway |
Cancer |
Mode of Regulation |
|
TXA2 pathway |
Lung cancer [18] |
Transcriptional regulation |
|
PGE2 pathway |
Neuroblastoma [39]; Colorectal cancer [17]; Lung cancer [41] |
Transcriptional regulation |
|
p53/miR-34/Nurr1 loop |
Colorectal cancer [40] |
Post-transcriptional regulation |
|
VEGF/PKD pathway |
Endothelia angiogenesis [33] |
Transcriptional regulation |
3.1. TXA2 Pathway
Aberrant TXA2-TP signaling axis has been reported in multiple cancers. TPα and TPβ are the two isoforms of TP and they couple to guanine nucleotide-binding protein (G protein) for functional mediation [42,43][40][41]. TPα couples to Gs and Gq, whereas TPβ couples to Gi and Gq [43][41]. Overexpression of TXA2, a COX-2-derived product, and activation of TP promotes cell survival and growth of melanoma, prostate and urothelial cancer [18,41,44,45][18][39][42][43].
The TXA2-TP signaling pathway can be elucidated as follows: I-BOP, a TP agonist, is found to induce Nurr1 expression in three ways [18][18]. (1) cAMP/protein kinase A (PKA)/CREB pathway: Upon TP activation, TPα couples to Gs to generate cAMP, which subsequently activates PKA and CREB; (2) ERK/CREB pathway: I-BOP induces MEK and ERK phosphorylation, possibly via Ras activation, leading to CREB activation; (3) PKC/CREB pathway: When TP is activated, TPβ links to phospholipase C (PLC) via coupling to Gq to activate protein kinase C (PKC), which further phosphorylates CREB. Meanwhile, PKC but not PKA is found to promote TP-mediated ERK1/2 phosphorylation, thereby suggesting that expression of I-BOP-induced Nurr1 may be partly due to activation of ERK by PKC.
3.2. PGE2 Pathway
PGE2 has been linked to cell proliferation, metastasis, cancer stemness, and chemoresistance in multiple cancers, including colorectal, endometrial, and lung cancer [46–49][43][44][45][46][47]. It mediates diverse and unique signaling networks via four G-protein-coupled receptors (EP1–EP4) for distinct second messenger pathways [17,39][17][39]. Similar to TXA2, PGE2 induces Nurr1 expression via cAMP-dependent and cAMP-independent pathways: (1) cAMP-dependent pathway: Upon activation by PGE2, EP2 couples to Gs, resulting in sequential cAMP activation, PKA activation, and CREB phosphorylation to initiate transcription of Nurr1 [18,49][18][47]; (2) cAMP-independent pathway: EP1 activated by PGE2 couples to G12/13, resulting in Rho activation [50][48]. Activated Rho then phosphorylates I-κB, leading to phosphorylation and dissociation of PKAc from the I-κB/NF-κB/PKAc complex and subsequent PKA-c-dependent phosphorylation of NF-κB and CREB to induce Nurr1 transcription[38] [39]. In addition, activated EP1 also couples to Gi to upregulate hypoxia-inducible factor-1 alpha (HIF1-α) via the PI3K/Akt/mTOR signaling pathway [51][49].
3.3. P53/miR-34/Nurr1 Loop
p53 is one of the most studied tumor suppressor proteins in cancer that stabilizes various genotoxic and cellular stresses, including DNA damage, oncogenic activation, hypoxia, and nutrient deprivation through transcription-dependent and -independent mechanisms [52,53][50][51]. Mounting evidence suggests that miRs are important components in the p53-dependent tumor suppressor network. MiR-34 (with isoforms miR34a and miR34b/c) is the direct transcriptional target of p53 and plays an important role in inducing apoptosis and cell cycle arrest by post-transcriptional regulation of miR-34 responsive genes [54–57][52][53][54][55]. MiR-34 has been shown to regulate and suppress Nurr1 expression by binding to the 3¢UTR of Nurr1 mRNA[40,57,58] [40][55][56]. Reduced Nurr1 protein expression mitigates its interaction with p53, thereby forming a tumor-suppressive p53/miR-34/Nurr1 loop. Nonetheless, when there is dysregulation of expression of any member in the loop, for instance, loss of p53 expression leads to decreased miR-34 expression and subsequent upregulation of Nurr1. This creates a positive feedback mechanism that shifts the regulatory loop from being tumor suppressive to oncogenic.
3.4. VEGF/Protein Kinase D (PKD) Pathway
Angiogenesis, a hallmark of cancer, is important for supplying nutrients to tumor masses for growth and metastasis [59][57]. These tumor blood vessels are characterized by immaturity and impaired functionality and offer survival advantages to tumor tissues, such as providing a hypoxic tumor microenvironment and reducing infiltration of immune cells [60][58]. The efficacy of radiotherapy, which relies on reactive oxygen species generation, and delivery of chemotherapeutic drugs have been hindered by hypoxic conditions and vessel immaturity, respectively [33][33]. Among various growth factors secreted by the tumor and stromal cells, VEGF plays a critical role in neovascularization. Evidence suggests that VEGF induces Nurr1 promoter activity and expression in endothelial cells. Silencing of Nurr1 suppresses endothelial cell proliferation, migration, and matrigel angiogenesis. VEGF-induced Nurr1 expression is mediated by VEGF receptor 2 (VEGFR2). It phosphorylates PKC, PKD, and CREB in a sequential manner [33,61,62][59][60]. Activated CREB then binds to the Nurr1 promoter to initiate transcription. In addition, VEGF also modulates NF-κB activation but with a moderate effect, as deletion of the upstream κB site moderately reduces Nurr1 promoter activity [33][33]. Taken together, VEGF induces Nurr1 expression primarily via a VEGF2-mediated PKC-dependent PKD axis.
References
- Giguere, V. Orphan nuclear receptors: From gene to function. Endocr. Rev. 1999, 20, 689–725, doi:10.1210/edrv.20.5.0378.
- Maxwell, M.A.; Muscat, G.E. The NR4A subgroup: Immediate early response genes with pleiotropic physiological roles. Nucl. Recept. Signal. 2006, 4, e002, doi:10.1621/nrs.04002.
- Beard, J.A.; Tenga, A.; Chen, T. The interplay of NR4A receptors and the oncogene-tumor suppressor networks in cancer. Cell. Signal. 2015, 27, 257–266, doi:10.1016/j.cellsig.2014.11.009.
- Sirin, O.; Lukov, G.L.; Mao, R.; Conneely, O.M.; Goodell, M.A. The orphan nuclear receptor Nurr1 restricts the proliferation of haematopoietic stem cells. Nat. Cell Biol. 2010, 12, 1213–1219, doi:10.1038/ncb2125.
- Saucedo-Cardenas, O.; Kardon, R.; Ediger, T.R.; Lydon, J.P.; Conneely, O.M. Cloning and structural organization of the gene encoding the murine nuclear receptor transcription factor, NURR1. Gene 1997, 187, 135–139.
- Zhao, Y.; Bruemmer, D. NR4A Orphan Nuclear Receptors in Cardiovascular Biology. Drug Discov. Today Dis. Mech. 2009, 6, e43–e48, doi:10.1016/j.ddmec.2009.06.001.
- Wang, Z.; Benoit, G.; Liu, J.; Prasad, S.; Aarnisalo, P.; Liu, X.; Xu, H.; Walker, N.P.; Perlmann, T. Structure and function of Nurr1 identifies a class of ligand-independent nuclear receptors. Nature 2003, 423, 555–560, doi:10.1038/nature01645.
- Jakaria, M.; Haque, M.E.; Cho, D.-Y.; Azam, S.; Kim, I.-S.; Choi, D.-K. Molecular Insights into NR4A2(Nurr1): An Emerging Target for Neuroprotective Therapy Against Neuroinflammation and Neuronal Cell Death. Mol. Neurobiol. 2019, 56, 5799–5814, doi:10.1007/s12035-019-1487-4.
- Bushue, N.; Wan, Y.J. Retinoid pathway and cancer therapeutics. Adv. Drug Deliv. Rev. 2010, 62, 1285–1298, doi:10.1016/j.addr.2010.07.003.
- Zagani, R.; Hamzaoui, N.; Cacheux, W.; de Reynies, A.; Terris, B.; Chaussade, S.; Romagnolo, B.; Perret, C.; Lamarque, D. Cyclooxygenase-2 inhibitors down-regulate osteopontin and Nr4A2-new therapeutic targets for colorectal cancers. Gastroenterology 2009, 137, 1358–1366, doi:10.1053/j.gastro.2009.06.039.
- Ke, N.; Claassen, G.; Yu, D.H.; Albers, A.; Fan, W.; Tan, P.; Grifman, M.; Hu, X.; Defife, K.; Nguy, V.; et al. Nuclear hormone receptor NR4A2 is involved in cell transformation and apoptosis. Cancer Res. 2004, 64, 8208–8212, doi:10.1158/0008-5472.Can-04-2134.
- Wang, J.; Yang, J.; Zou, Y.; Huang, G.L.; He, Z.W. Orphan nuclear receptor nurr1 as a potential novel marker for progression in human prostate cancer. Asian Pac. J. Cancer Prev. 2013, 14, 2023–2028, doi:10.7314/apjcp.2013.14.3.2023.
- Ji, L.; Gong, C.; Ge, L.; Song, L.; Chen, F.; Jin, C.; Zhu, H.; Zhou, G. Orphan nuclear receptor Nurr1 as a potential novel marker for progression in human pancreatic ductal adenocarcinoma. Exp. Ther. Med. 2017, 13, 551–559, doi:10.3892/etm.2016.3968.
- Han, Y.; Cai, H.; Ma, L.; Ding, Y.; Tan, X.; Chang, W.; Guan, W.; Liu, Y.; Shen, Q.; Yu, Y.; et al. Expression of orphan nuclear receptor NR4A2 in gastric cancer cells confers chemoresistance and predicts an unfavorable postoperative survival of gastric cancer patients with chemotherapy. Cancer 2013, 119, 3436–3445, doi:10.1002/cncr.28228.
- Han, Y.; Cai, H.; Ma, L.; Ding, Y.; Tan, X.; Liu, Y.; Su, T.; Yu, Y.; Chang, W.; Zhang, H.; et al. Nuclear orphan receptor NR4A2 confers chemoresistance and predicts unfavorable prognosis of colorectal carcinoma patients who received postoperative chemotherapy. Eur J. Cancer 2013, 49, 3420–3430, doi:10.1016/j.ejca.2013.06.001.
- Kok-Ting Wan, P.; Ho-Yin Leung, T.; Kwan-Yee Siu, M.; Mo, X.; Wai-Man Tang, H.; Kar-Loen Chan, K.; Nga-Yin Cheung, A.; Yuen-Sheung Ngan, H. HPV-induced Nurr1 promotes cancer aggressiveness, self-renewal, and radioresistance via ERK and AKT signaling in cervical cancer. Cancer Lett 2020, doi:10.1016/j.canlet.2020.09.025.
- Holla, V.R.; Mann, J.R.; Shi, Q.; DuBois, R.N. Prostaglandin E2 regulates the nuclear receptor NR4A2 in colorectal cancer. J. Biol. Chem. 2006, 281, 2676–2682, doi:10.1074/jbc.M507752200.
- Li, X.; Tai, H.H. Activation of thromboxane A(2) receptors induces orphan nuclear receptor Nurr1 expression and stimulates cell proliferation in human lung cancer cells. Carcinogenesis 2009, 30, 1606–1613, doi:10.1093/carcin/bgp161.
- Inamoto, T.; Czerniak, B.A.; Dinney, C.P.; Kamat, A.M. Cytoplasmic mislocalization of the orphan nuclear receptor Nurr1 is a prognostic factor in bladder cancer. Cancer 2010, 116, 340–346, doi:10.1002/cncr.24737.
- Misund, K.; Selvik, L.K.; Rao, S.; Norsett, K.; Bakke, I.; Sandvik, A.K.; Laegreid, A.; Bruland, T.; Prestvik, W.S.; Thommesen, L. NR4A2 is regulated by gastrin and influences cellular responses of gastric adenocarcinoma cells. PLoS ONE 2013, 8, e76234, doi:10.1371/journal.pone.0076234.
- Maruyama, K.; Tsukada, T.; Bandoh, S.; Sasaki, K.; Ohkura, N.; Yamaguchi, K. Retinoic acids differentially regulate NOR-1 and its closely related orphan nuclear receptor genes in breast cancer cell line MCF-7. Biochem. Biophys. Res. Commun. 1997, 231, 417–420, doi:10.1006/bbrc.1997.6122.
- Llopis, S.; Singleton, B.; Duplessis, T.; Carrier, L.; Rowan, B.; Williams, C. Dichotomous roles for the orphan nuclear receptor NURR1 in breast cancer. Bmc Cancer 2013, 13, 139, doi:10.1186/1471-2407-13-139.
- Inamoto, T.; Papineni, S.; Chintharlapalli, S.; Cho, S.D.; Safe, S.; Kamat, A.M. 1,1-Bis(3'-indolyl)-1-(p-chlorophenyl)methane activates the orphan nuclear receptor Nurr1 and inhibits bladder cancer growth. Mol. Cancer Ther. 2008, 7, 3825–3833, doi:10.1158/1535-7163.Mct-08-0730.
- Chang, W.; Ma, L.; Lin, L.; Gu, L.; Liu, X.; Cai, H.; Yu, Y.; Tan, X.; Zhai, Y.; Xu, X.; et al. Identification of novel hub genes associated with liver metastasis of gastric cancer. Int. J. Cancer 2009, 125, 2844–2853, doi:10.1002/ijc.24699.
- Sun, L.; Liu, M.; Sun, G.C.; Yang, X.; Qian, Q.; Feng, S.; Mackey, L.V.; Coy, D.H. Notch Signaling Activation in Cervical Cancer Cells Induces Cell Growth Arrest with the Involvement of the Nuclear Receptor NR4A2. J. Cancer 2016, 7, 1388–1395, doi:10.7150/jca.15274.
- Karki, K.; Li, X.; Jin, U.-H.; Mohankumar, K.; Zarei, M.; Michelhaugh, S.K.; Mittal, S.; Tjalkens, R.; Safe, S. Nuclear receptor 4A2 (NR4A2) is a druggable target for glioblastomas. J. Neuro Oncol. 2020, 146, 25–39, doi:10.1007/s11060-019-03349-y.
- Lin, B.; Kolluri, S.K.; Lin, F.; Liu, W.; Han, Y.H.; Cao, X.; Dawson, M.I.; Reed, J.C.; Zhang, X.K. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell 2004, 116, 527–540.
- Wang, D.; DuBois, R.N. An inflammatory mediator, prostaglandin E2, in colorectal cancer. Cancer J. 2013, 19, 502–510, doi:10.1097/ppo.0000000000000003.
- Norregaard, R.; Kwon, T.H.; Frokiaer, J. Physiology and pathophysiology of cyclooxygenase-2 and prostaglandin E2 in the kidney. Kidney Res. Clin. Pract. 2015, 34, 194–200, doi:10.1016/j.krcp.2015.10.004.
- Likui, W.; Hong, W.; Shuwen, Z. Clinical significance of the upregulated osteopontin mRNA expression in human colorectal cancer. J. Gastrointest. Surg. 2010, 14, 74–81, doi:10.1007/s11605-009-1035-z.
- Agrawal, D.; Chen, T.; Irby, R.; Quackenbush, J.; Chambers, A.F.; Szabo, M.; Cantor, A.; Coppola, D.; Yeatman, T.J. Osteopontin identified as lead marker of colon cancer progression, using pooled sample expression profiling. J. Natl. Cancer Inst. 2002, 94, 513–521, doi:10.1093/jnci/94.7.513.
- Han, Y.F.; Cao, G.W. Role of nuclear receptor NR4A2 in gastrointestinal inflammation and cancers. World J. Gastroenterol. 2012, 18, 6865–6873, doi:10.3748/wjg.v18.i47.6865.
- Zhao, D.; Desai, S.; Zeng, H. VEGF stimulates PKD-mediated CREB-dependent orphan nuclear receptor Nurr1 expression: Role in VEGF-induced angiogenesis. Int. J. Cancer 2011, 128, 2602–2612, doi:10.1002/ijc.25600.
- Lin, P.-C.; Chen, Y.-L.; Chiu, S.-C.; Yu, Y.-L.; Chen, S.-P.; Chien, M.-H.; Chen, K.-Y.; Chang, W.-L.; Lin, S.-Z.; Chiou, T.-W.; et al. Orphan nuclear receptor, Nurr-77 was a possible target gene of butylidenephthalide chemotherapy on glioblastoma multiform brain tumor. J. Neurochem. 2008, 106, 1017–1026, doi:10.1111/j.1471-4159.2008.05432.x.
- Mullican, S.E.; Zhang, S.; Konopleva, M.; Ruvolo, V.; Andreeff, M.; Milbrandt, J.; Conneely, O.M. Abrogation of nuclear receptors Nr4a3 andNr4a1 leads to development of acute myeloid leukemia. Nat. Med. 2007, 13, 730–735, doi:10.1038/nm1579.
- Ramirez-Herrick, A.M.; Mullican, S.E.; Sheehan, A.M.; Conneely, O.M. Reduced NR4A gene dosage leads to mixed myelodysplastic/myeloproliferative neoplasms in mice. Blood 2011, 117, 2681–2690, doi:10.1182/blood-2010-02-267906.
- Beard, J.A.; Tenga, A.; Hills, J.; Hoyer, J.D.; Cherian, M.T.; Wang, Y.D.; Chen, T. The orphan nuclear receptor NR4A2 is part of a p53-microRNA-34 network. Sci. Rep. 2016, 6, 25108, doi:10.1038/srep25108.
- Ji, R.; Sanchez, C.M.; Chou, C.L.; Chen, X.B.; Woodward, D.F.; Regan, J.W. Prostanoid EP(1) receptors mediate up-regulation of the orphan nuclear receptor Nurr1 by cAMP-independent activation of protein kinase A, CREB and NF-kappaB. Br. J. Pharm. 2012, 166, 1033–1046, doi:10.1111/j.1476-5381.2011.01817.x.
- Shimizu, T.; Fujii, T.; Takahashi, Y.; Takahashi, Y.; Suzuki, T.; Ukai, M.; Tauchi, K.; Horikawa, N.; Tsukada, K.; Sakai, H. Up-regulation of Kv7.1 channels in thromboxane A2-induced colonic cancer cell proliferation. Pflug. Arch. 2014, 466, 541–548, doi:10.1007/s00424-013-1341-x.
- Young, M.R.; Young, M.E.; Lozano, Y.; Coogan, M.; Bagash, J.M. Regulation of protein kinase A activation and prostaglandin E2-stimulated migration of Lewis lung carcinoma clones. Int. J. Cancer 1991, 49, 150–155.
- Hirata, T.; Ushikubi, F.; Kakizuka, A.; Okuma, M.; Narumiya, S. Two thromboxane A2 receptor isoforms in human platelets. Opposite coupling to adenylyl cyclase with different sensitivity to Arg60 to Leu mutation. J. Clin. Investig. 1996, 97, 949–956, doi:10.1172/jci118518.
- Sobolesky, P.M.; Halushka, P.V.; Garrett-Mayer, E.; Smith, M.T.; Moussa, O. Regulation of the tumor suppressor FOXO3 by the thromboxane-A2 receptors in urothelial cancer. PLoS ONE 2014, 9, e107530, doi:10.1371/journal.pone.0107530.
- Keating, G.L.; Reid, H.M.; Eivers, S.B.; Mulvaney, E.P.; Kinsella, B.T. Transcriptional regulation of the human thromboxane A2 receptor gene by Wilms' tumor (WT)1 and hypermethylated in cancer (HIC) 1 in prostate and breast cancers. Biochim. Biophys. Acta 2014, 1839, 476–492, doi:10.1016/j.bbagrm.2014.04.010.
- Ke, J.; Yang, Y.; Che, Q.; Jiang, F.; Wang, H.; Chen, Z.; Zhu, M.; Tong, H.; Zhang, H.; Yan, X.; et al. Prostaglandin E2 (PGE2) promotes proliferation and invasion by enhancing SUMO-1 activity via EP4 receptor in endometrial cancer. Tumor Biol. 2016, 37, 12203–12211, doi:10.1007/s13277-016-5087-x.
- Park, Y.R.; Seo, S.Y.; Kim, S.L.; Zhu, S.M.; Chun, S.; Oh, J.M.; Lee, M.R.; Kim, S.H.; Kim, I.H.; Lee, S.O.; et al. MiRNA-206 suppresses PGE2-induced colorectal cancer cell proliferation, migration, and invasion by targetting TM4SF1. Biosci. Rep. 2018, 38, doi:10.1042/bsr20180664.
- Nakanishi, M.; Rosenberg, D.W. Multifaceted roles of PGE2 in inflammation and cancer. Semin. Immunopathol. 2013, 35, 123–137, doi:10.1007/s00281-012-0342-8.
- Wang, D.; Fu, L.; Sun, H.; Guo, L.; DuBois, R.N. Prostaglandin E2 Promotes Colorectal Cancer Stem Cell Expansion and Metastasis in Mice. Gastroenterology 2015, 149, 1884–1895, doi:10.1053/j.gastro.2015.07.064.
- Juneja, J.; Casey, P.J. Role of G12 proteins in oncogenesis and metastasis. Br. J. Pharm. 2009, 158, 32–40, doi:10.1111/j.1476-5381.2009.00180.x.
- Ji, R.; Chou, C.L.; Xu, W.; Chen, X.B.; Woodward, D.F.; Regan, J.W. EP1 prostanoid receptor coupling to G i/o up-regulates the expression of hypoxia-inducible factor-1 alpha through activation of a phosphoinositide-3 kinase signaling pathway. Mol. Pharmacol. 2010, 77, 1025–1036, doi:10.1124/mol.110.063933.
- Hong, B.; van den Heuvel, A.P.; Prabhu, V.V.; Zhang, S.; El-Deiry, W.S. Targeting tumor suppressor p53 for cancer therapy: Strategies, challenges and opportunities. Curr. Drug Targets 2014, 15, 80–89.
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur J. Cancer 2017, 83, 258–265, doi:10.1016/j.ejca.2017.06.023.
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134, doi:10.1038/nature05939.
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 2007, 26, 731–743, doi:10.1016/j.molcel.2007.05.017.
- Hermeking, H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010, 17, 193–199, doi:10.1038/cdd.2009.56.
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752, doi:10.1016/j.molcel.2007.05.010.
- Zhang, T.; Wang, P.; Ren, H.; Fan, J.; Wang, G. NGFI-B nuclear orphan receptor Nurr1 interacts with p53 and suppresses its transcriptional activity. Mol. Cancer Res. 2009, 7, 1408–1415, doi:10.1158/1541-7786.Mcr-08-0533.
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307, doi:10.1038/nature10144.
- Viallard, C.; Larrivee, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426, doi:10.1007/s10456-017-9562-9.
- Zeng, H.; Sanyal, S.; Mukhopadhyay, D. Tyrosine residues 951 and 1059 of vascular endothelial growth factor receptor-2 (KDR) are essential for vascular permeability factor/vascular endothelial growth factor-induced endothelium migration and proliferation, respectively. J. Biol. Chem. 2001, 276, 32714–32719, doi:10.1074/jbc.M103130200.
- Gille, H.; Kowalski, J.; Li, B.; LeCouter, J.; Moffat, B.; Zioncheck, T.F.; Pelletier, N.; Ferrara, N. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J. Biol. Chem. 2001, 276, 3222–3230, doi:10.1074/jbc.M002016200.