Pulmonary fibrosis (PF) is the end-stage consequence of various interstitial lung diseases (ILD). It is a chronic progressive disease with an average survival of 3–5 years after diagnosis. The pathological features of PF are the abnormal activation and proliferation of myofibroblasts and the extraordinary deposition of the extracellular matrix (ECM).
- pulmonary fibrosis
- endothelial cells
- myofibroblasts
1. Introduction
Pulmonary fibrosis (PF) is the end-stage consequence of various interstitial lung diseases (ILD). It is a chronic progressive disease with an average survival of 3–5 years after diagnosis [1]. PF results from the dysregulation of alveolar epithelial cell (AECs) repair in response to alveolar and vascular damage, which leads to the excessive accumulation of ECM, proliferation of myofibroblasts, distortion of pulmonary architecture, and loss of pulmonary tissue function [2]. Studies have reported that circulating fibrocytes, pulmonary alveolar epithelial cells, fibroblasts, pericytes, macrophages, and endothelial cells were the progenitors of myofibroblasts and contributed to the development of PF.
2. Etiology of Common Pulmonary Fibrosis
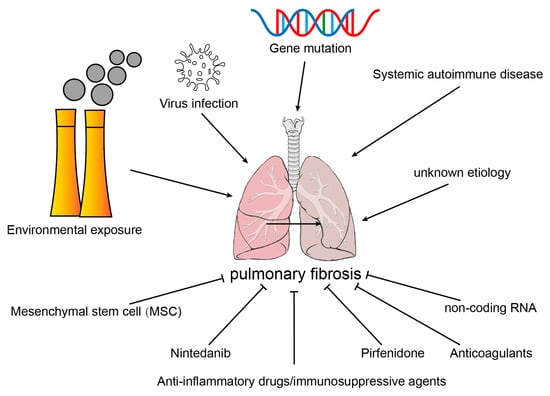
2.1. Environmental Exposure
2.2. Virus Infection
2.3. Systemic Autoimmune Disease
2.4. Gene Mutation
2.5. Idiopathic Pulmonary Fibrosis
3. Source of Myofibroblasts in Fibrotic Lung Tissues
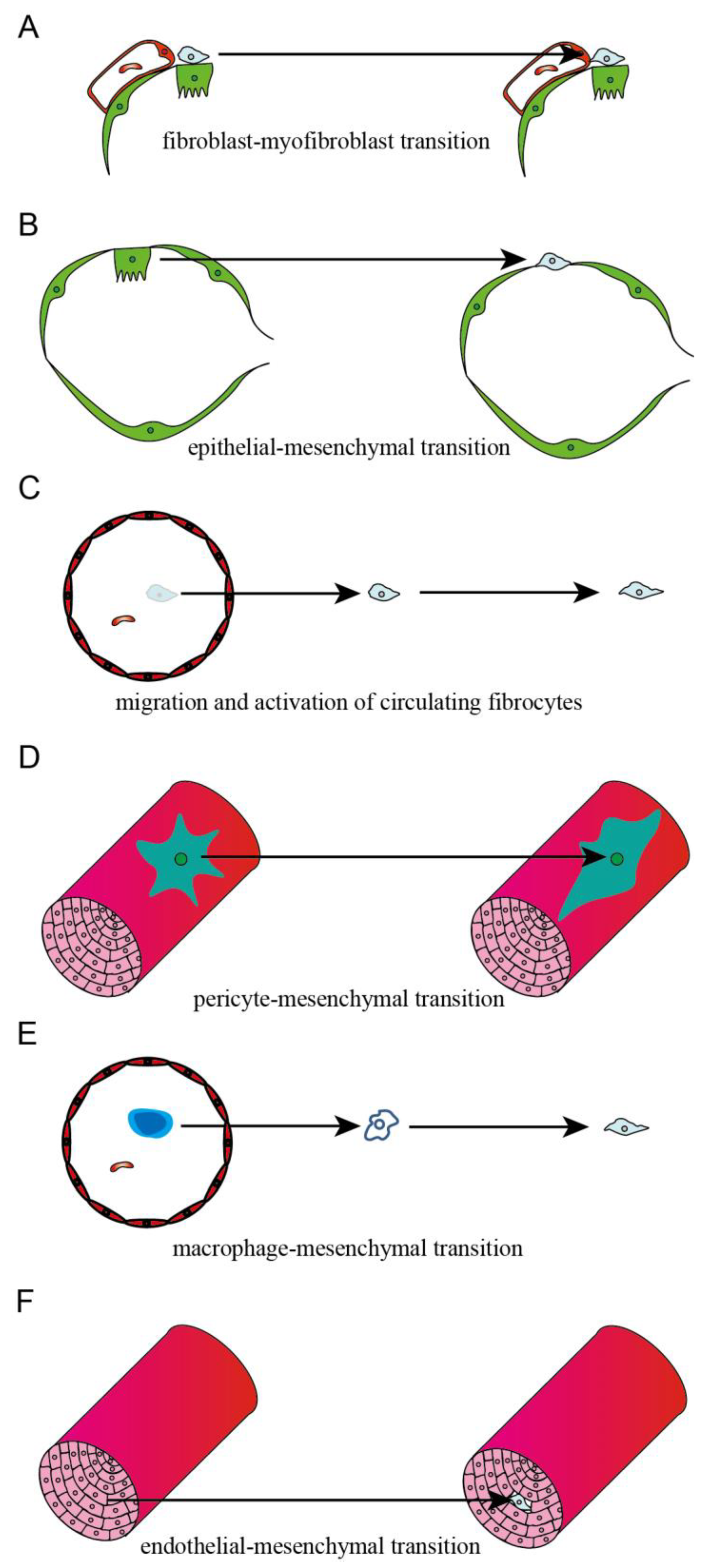
3.1. Interstitial Fibroblast
3.2. Epithelial Cell
3.3. Circulating Fibrocyte
3.4. Microvascular Pericyte
3.5. Macrophage
3.6. Endothelial Cell
References
- Cheng, L.; Wang, D.; Deng, B.; Li, J.; Zhang, J.; Guo, X.; Yan, T.; Yue, X.; An, Y.; Zhang, B.; et al. DR7dA, a Novel Antioxidant Peptide Analog, Demonstrates Antifibrotic Activity in Pulmonary Fibrosis In Vivo and In Vitro. J. Pharmacol. Exp. Ther. 2022, 382, 100–112.
- Marchioni, A.; Tonelli, R.; Cerri, S.; Castaniere, I.; Andrisani, D.; Gozzi, F.; Bruzzi, G.; Manicardi, L.; Moretti, A.; Demurtas, J.; et al. Pulmonary Stretch and Lung Mechanotransduction: Implications for Progression in the Fibrotic Lung. Int. J. Mol. Sci. 2021, 22, 6443.
- Fujimoto, H.; Kobayashi, T.; Azuma, A. Idiopathic Pulmonary Fibrosis: Treatment and Prognosis. Clin. Med. Insights Circ. Respir. Pulm. Med. 2015, 9, 179–185.
- Somogyi, V.; Chaudhuri, N.; Torrisi, S.E.; Kahn, N.; Muller, V.; Kreuter, M. The therapy of idiopathic pulmonary fibrosis: What is next? Eur. Respir. Rev. 2019, 28, 153.
- Gao, Y.; Sun, J.; Dong, C.; Zhao, M.; Hu, Y.; Jin, F. Extracellular Vesicles Derived from Adipose Mesenchymal Stem Cells Alleviate PM2.5-Induced Lung Injury and Pulmonary Fibrosis. Med. Sci. Monit. 2020, 26, e922782.
- Xu, M.; Wang, X.; Xu, L.; Zhang, H.; Li, C.; Liu, Q.; Chen, Y.; Chung, K.F.; Adcock, I.M.; Li, F. Chronic lung inflammation and pulmonary fibrosis after multiple intranasal instillation of PM2.5 in mice. Environ. Toxicol. 2021, 36, 1434–1446.
- Cheresh, P.; Morales-Nebreda, L.; Kim, S.J.; Yeldandi, A.; Williams, D.B.; Cheng, Y.; Mutlu, G.M.; Budinger, G.R.; Ridge, K.; Schumacker, P.T.; et al. Asbestos-induced pulmonary fibrosis is augmented in 8-oxoguanine DNA glycosylase knockout mice. Am. J. Respir. Cell Mol. Biol. 2015, 52, 25–36.
- Sheng, G.; Chen, P.; Wei, Y.; Yue, H.; Chu, J.; Zhao, J.; Wang, Y.; Zhang, W.; Zhang, H.L. Viral Infection Increases the Risk of Idiopathic Pulmonary Fibrosis: A Meta-Analysis. Chest 2020, 157, 1175–1187.
- Huang, W.J.; Tang, X.X. Virus infection induced pulmonary fibrosis. J. Transl. Med. 2021, 19, 496.
- Fukui, Y.; Nakamura, K.; Hirabayashi, M.; Miyagawa, T.; Toyama, S.; Omatsu, J.; Awaji, K.; Ikawa, T.; Norimatsu, Y.; Yoshizaki, A.; et al. Serum vasohibin-1 levels: A potential marker of dermal and pulmonary fibrosis in systemic sclerosis. Exp. Dermatol. 2021, 30, 951–958.
- Wang, S.; Liu, M.; Li, X.; Zhang, J.; Wang, F.; Zhang, C.; Roden, A.; Ryu, J.H.; Warrington, K.J.; Sun, J.; et al. Canonical and noncanonical regulatory roles for JAK2 in the pathogenesis of rheumatoid arthritis-associated interstitial lung disease and idiopathic pulmonary fibrosis. FASEB J. 2022, 36, e22336.
- Shen, N.; Zhou, X.; Jin, X.; Lu, C.; Hu, X.; Zhang, Y.; Jiang, Y.; Xu, Q.; Xu, X.; Liu, M.; et al. MDA5 expression is associated with TGF-beta-induced fibrosis: Potential mechanism of interstitial lung disease in anti-MDA5 dermatomyositis. Rheumatology 2022, 62, 373–383.
- Shi, L.; Fu, Q.; Chen, N.; Liu, R.; Zheng, Y. Angiopoietin-like protein 2 as a novel marker for patients with primary Sjogren’s syndrome-related interstitial lung disease. Clin. Exp. Med. 2020, 20, 393–399.
- Caballero, I.; Ringot-Destrez, B.; Si-Tahar, M.; Barbry, P.; Guillon, A.; Lantier, I.; Berri, M.; Chevaleyre, C.; Fleurot, I.; Barc, C.; et al. Evidence of early increased sialylation of airway mucins and defective mucociliary clearance in CFTR-deficient piglets. J. Cyst. Fibros. 2021, 20, 173–182.
- Pardo, A.; Selman, M. The Interplay of the Genetic Architecture, Aging, and Environmental Factors in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 64, 163–172.
- Lim, M.J.; Ahn, J.; Yi, J.Y.; Kim, M.H.; Son, A.R.; Lee, S.L.; Lim, D.S.; Kim, S.S.; Kang, M.A.; Han, Y.; et al. Induction of galectin-1 by TGF-beta1 accelerates fibrosis through enhancing nuclear retention of Smad2. Exp. Cell Res. 2014, 326, 125–135.
- Yao, L.; Conforti, F.; Hill, C.; Bell, J.; Drawater, L.; Li, J.; Liu, D.; Xiong, H.; Alzetani, A.; Chee, S.J.; et al. Paracrine signalling during ZEB1-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in lung fibrosis. Cell Death Differ. 2019, 26, 943–957.
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483.
- Chiang, H.Y.; Chu, P.H.; Lee, T.H. R1R2 peptide ameliorates pulmonary fibrosis in mice through fibrocyte migration and differentiation. PLoS ONE 2017, 12, e0185811.
- Aono, Y.; Kishi, M.; Yokota, Y.; Azuma, M.; Kinoshita, K.; Takezaki, A.; Sato, S.; Kawano, H.; Kishi, J.; Goto, H.; et al. Role of platelet-derived growth factor/platelet-derived growth factor receptor axis in the trafficking of circulating fibrocytes in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2014, 51, 793–801.
- Rowley, J.E.; Johnson, J.R. Pericytes in chronic lung disease. Int. Arch. Allergy Immunol. 2014, 164, 178–188.
- Sava, P.; Ramanathan, A.; Dobronyi, A.; Peng, X.; Sun, H.; Ledesma-Mendoza, A.; Herzog, E.L.; Gonzalez, A.L. Human pericytes adopt myofibroblast properties in the microenvironment of the IPF lung. JCI Insight 2017, 2, 24.
- Wang, Y.C.; Xie, H.; Zhang, Y.C.; Meng, Q.H.; Xiong, M.M.; Jia, M.W.; Peng, F.; Tang, D.L. Exosomal miR-107 antagonizes profibrotic phenotypes of pericytes by targeting a pathway involving HIF-1alpha/Notch1/PDGFRbeta/YAP1/Twist1 axis in vitro. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H520–H534.
- Nikolic-Paterson, D.J.; Wang, S.; Lan, H.Y. Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int. Suppl. 2014, 4, 34–38.
- Yang, F.; Chang, Y.; Zhang, C.; Xiong, Y.; Wang, X.; Ma, X.; Wang, Z.; Li, H.; Shimosawa, T.; Pei, L.; et al. UUO induces lung fibrosis with macrophage-myofibroblast transition in rats. Int. Immunopharmacol. 2021, 93, 107396.
- Hashimoto, N.; Phan, S.H.; Imaizumi, K.; Matsuo, M.; Nakashima, H.; Kawabe, T.; Shimokata, K.; Hasegawa, Y. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 43, 161–172.
- Gaikwad, A.V.; Lu, W.; Dey, S.; Bhattarai, P.; Chia, C.; Larby, J.; Haug, G.; Myers, S.; Jaffar, J.; Westall, G.; et al. Vascular remodelling in idiopathic pulmonary fibrosis patients and its detrimental effect on lung physiology: Potential role of endothelial-to-mesenchymal transition. ERJ Open Res. 2022, 8, 1.