Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Mariusz Mojzych and Version 2 by Rita Xu.
The Corey-Seebach reagent plays an important role in organic synthesis because of its broad synthetic applications. The Corey-Seebach reagent is formed by the reaction of an aldehyde or a ketone with 1,3-propane-dithiol under acidic conditions, followed by deprotonation with
n-butyllithium. A large variety of natural products (alkaloids, terpenoids, and polyketides) can be accessed successfully by utilizing this reagent.
-butyllithium. A large variety of natural products (alkaloids, terpenoids, and polyketides) can be accessed successfully by utilizing this reagent.
- Corey-Seebach reagent
- natural products
- alkaloids
1. Introduction
Elias James Corey is an American chemist well-known for his contribution to the development of methodology and theory of organic synthesis, especially retrosynthetic analysis. He was awarded a Nobel Prize in 1990 for the development of retrosynthetic analysis. His research cooperation with other famous organic chemists has resulted in various name reactions, based on his name, in organic chemistry [1]. One of his famous reactions is the Corey-Seebach reaction which was a combined work of Corey and Dieter Seebach (a German chemist). The Corey-Seebach reagent is formed by the reaction of an aldehyde or a ketone with 1,3-propane-dithiol in the presence of acidic conditions (Lewis acid). Corey-Seebach is a nucleophilic moiety and has widespread applications in various organic transformations. This reaction was first published in 1965, which reported the synthesis of dicarbonyl derivative from 1,3-dithiane [2]. This acyl anion intermediate easily provides access to α-hydroxy ketones [3][4][5][6][3,4,5,6] by reacting with a range of electrophiles, including carbonyl compounds (Figure 1) [7].
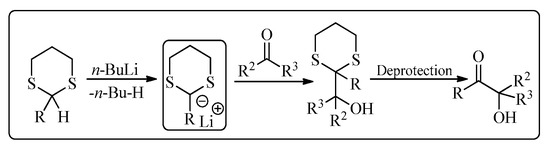
Figure 1. A typical Corey-Seebach reaction.
In order to regenerate the carbonyl group that was initially masked when dithiane was utilized as an acyl anion equivalent, it must be hydrolyzed at some point during synthesis. Deprotection has frequently been challenging to accomplish, especially for complicated and sensitive compounds, and as a result, numerous processes have been adopted. The use of traditional methods such as metal salts (mercury(II) chloride [8]) for the deprotection of 1,3-dithiane requires toxic reagents that are generally harmful to the environment. However, there are some facile and efficient methods available in the literature, i.e., 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) deprotection [9] and the use of iodine catalyst/H2O2 [10] which are more environment friendly.
In a typical 1,3-dithiane addition process, 1,3-dithiane is combined with an equimolar quantity of a strong base, such as n-butyllithium, and the resultant 2-lithio-1,3-dithiane should serve as an appropriate nucleophile. According to a different procedure described by Andersen et al. [11] the 1,3-dithiane equivalent, 2-trimethylsilyl-1,3-dithiane (TMS-dithiane), could be activated by a stoichiometric quantity of tetrabutylammonium fluoride (TBAF), resulting in the matching carbanion. Corey et al. claim that various cesium salt mixtures that include cesium fluoride may be used as heterogeneous desilylating reagents. There are just a few cases when TMS-dithiane has been activated catalytically, and most of these reactions involve the use of fluoride reagents in equimolar amounts [12].
The Corey-Seebach umpolung technique has been extensively utilized to manufacture a wide variety of natural products such as Swinholide A [13] (1, a marine natural product, derived from sponge Theonella swinhoei, which shows antitumor and antifungal activity), pironetin [14][15][16][14,15,16] (2, derived from Streptomyces fermentation broths, which exhibits plant growth regulating action) (Figure 2), ciguatoxin 1B [17] (3, one of the main toxins responsible for ciguatera fish poisoning (Figure 3), discovered from moray eel Gymnothorax javanicus), and maytansine [18][19][18,19] (4, shows antitumor activity) (Figure 4). Many synthetic [20] compounds, such as photolabile safety benzoin linkers [21] and bis-3,4-dihydroisoquinolium salts [22], have also been attained using the Corey-Seebach reagent. Earlier, Foubelo et al. published a review article in 2003 concerning the use of 1,3-dithianes in the synthesis of natural products [23]. Until now, the Corey-Seebach reagent has found valuable applications in organic synthesis. TheOur researchview article focuses on the utilization of Corey-Seebach reagent in the synthesis of noteworthy natural and synthetic organic compounds reported post-2006.
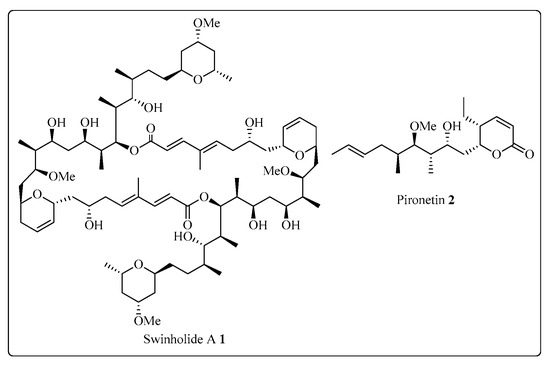
Figure 2. Structure of Swinholide A 1 and pironetin 2.
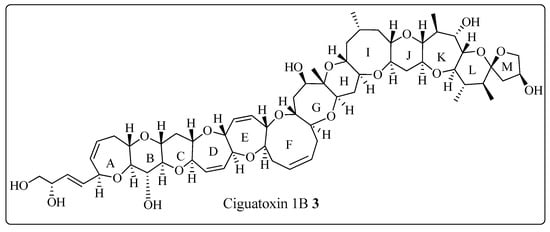
Figure 3. Structure of ciguatoxin 1B 3.
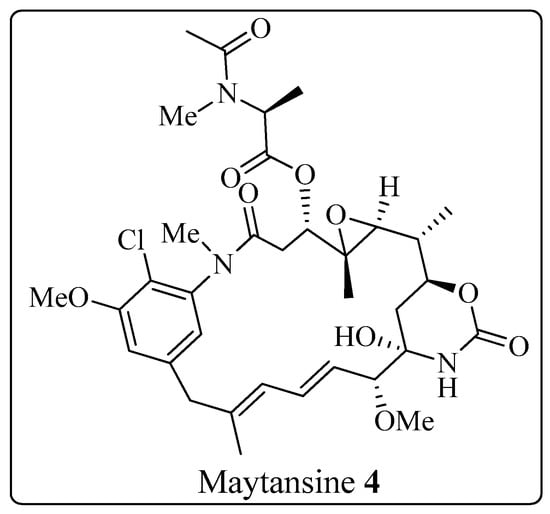
Figure 4. Structure of maytansine 4.
2. Alkaloid-Based Natural Products Synthesis
2.1. Lycoplanine A Alkaloids
Lycopodium alkaloids are known to play an effective role in the medication of Alzheimer’s disease [24][25][26][24,25,26]. Over 300 lycopodium alkaloids have so far been isolated, and a number of total syntheses of these alkaloids have been published [27][28][29][27,28,29]. In 2017, Zhao and co-workers [30] first isolated lycoplanine A, a lycopodium alkaloid with the γ-lactone ring. According to biological investigations, lycoplanine alkaloid is a strong inhibitor of the calcium channel (Cav3.1 T-type) with an IC50 value of 6.06 μM. In 2021, Gao et al. [31] reported the synthesis of lycoplanine A isomer by utilizing the Corey-Seebach reagent. To achieve this task, the C=C bond was introduced by using 1,4-dithiane 5 and crotonaldehyde (E/Z > 98%) to afford alcohol 6 in an 88% yield, followed by oxidation to provide the product 7 with an 80% yield. Compound 7 was then treated with Nysted reagent for the introduction of the second C=C, followed by the introduction of fragment A to afford compound 8 by using the Mitsunobu reaction. After a few steps, compound 9 was formed, which upon reaction with Crabtree’s catalyst provided a tetrasubstituted C=C bond product 10 with excellent stereo and regioselectivities. After deprotection of the Boc group, compound 10 was immediately exposed to AcOH, initiating a cascade reaction that produced the stereo-specific cyclized product 11 with a 35% yield. The deprotection of the thioketal group was achieved by using PIFA to afford lycoplanine A 12 isomer with an 83% yield (Scheme 1).
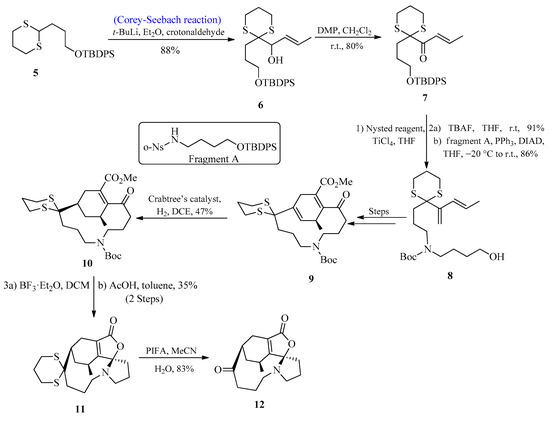
Scheme 1. Synthesis of lycoplanine A 12 isomer via Corey-Seebach reagent.
2.2. Diterpenoid Alkaloids
Diterpenoid alkaloids have been the focus of study by scientists all over the globe because of their fascinating bioactivities and complicated structures [32]. These biologically active compounds were extracted from Delphinium and Aconitum species that belong to the Ranunculaceae family [33]. In 2017, Min Zhu et al. [34] reported the synthesis of hetidine-type C20-diterpenoid alkaloids by utilizing the Corey-Seebach reagent as a key step. For this purpose, 2-lithio-1,3-dithiane species 14 were reacted with iodide 13, followed by the deprotection of methoxymethyl to afford olefinic phenol 15 with an 80% yield. In the next step, compound 15 was treated with PhI(OAc)2, followed by the addition of Sml2 to obtain compound 16, which could then be transformed into the desired hetidine-type diterpenoid alkaloid 17 after a series of reactions (Scheme 2).
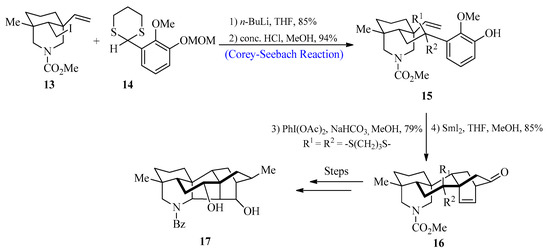
Scheme 2. Synthesis of hetidine-based diterpenoid 17 alkaloid.
3. Terpenoids-Based Natural Products Synthesis
3.1. Bisnorditerpene
Diterpenoids are important natural products that display a wide range of chemical diversity and are useful both in medicine and industry. A large number of known diterpenoid compounds are isolated from plants and fungi, and investigations into these species have provided an understanding of their production [35]. In 2010, Pessoa et al. [36] first isolated a bisnorditerpene from Croton regelianus var. matosii. This herb is utilized in traditional medicine in the Northeastern state of “Caatinga”. In 2016, Xu et al. [37] designed a new strategy for the synthesis of bisnorditerpene by utilizing the Corey-Seebach reagent. To achieve this, an aldehyde 18 was allowed to react with 1,3-propane-dithiol 19 to furnish dithiane 20, which upon lithiation with epoxide [38] 21 by using the Corey-Seebach reaction afforded precursor 22 followed by the addition of Lewis acid to obtain tricyclic alcohol 23 with a 55% yield. In the next step, secondary alcohol 24 was obtained by desulfurization of 23 with Raney-Ni, followed by the oxidation of 24 with Dess-Martin periodinane (DMP) to afford ketone 25 with a 95% yield. The final process involved the demethylation of ketone 25 with BBr3 along with the addition of Phl(OAc)2 in CH3CN to generate bisnorditerpene 26 (Scheme 3).
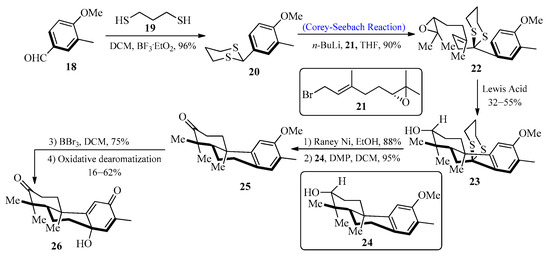
Scheme 3. Synthesis of bisnorditerpene 26.
3.2. Totarol Synthesis
Totarol belongs to diterpenes that are found in the sap of podocarpus totara, a New Zealand native conifer [39]. The antimicrobial properties [40][41][42][43][40,41,42,43] of the secondary metabolites in this sap are well known. The wood of this tree displays resistance against rot. Toothpaste and acne medications are just a couple of consumer goods that can contain totarol as an antibacterial ingredient. In 2010, Kim et al. [44] synthesized totarol by utilizing the Corey-Seebach approach as an important key step. The goal of their research was to synthesize totarol diterpenes as a part of a larger research project to determine the mechanism by which tiny molecules could inactivate FtsZ. In order to achieve this, benzonitrile 27 was treated with i-PrMgCl to produce compound 28, followed by the reduction and thioacetal formation to obtain product 29. In the next step, alkene 30 was synthesized through lithiation of 29 with fragment B followed by alkylation, respectively. Treatment of compound 30 with AD-mix-β afforded regio-isomeric diol 31 with 90–95% enantiomeric excess, and after a few steps, totarolone 32 was formed with a 33% yield. Totarolone 32 was transformed into the desired totarol 33 through the Wolff-Kishner reduction (Scheme 4).
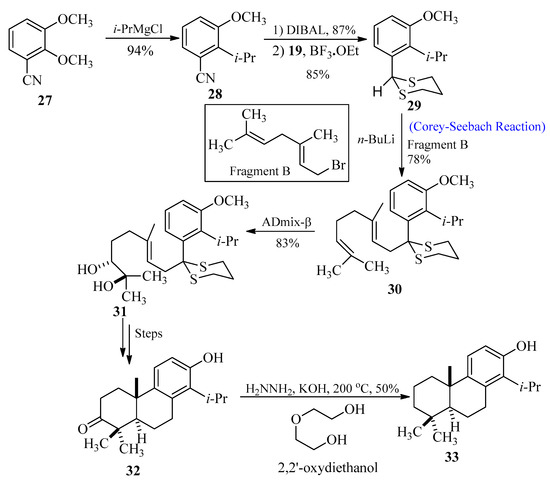
Scheme 4. Synthesis of totarol 33 via Corey-Seebach approach.
4. Polyketide Based Natural Products
4.1. Ambruticin J Synthesis
A significant class of polyketide-based natural compounds known as ambruticin was initially isolated from the bacterium Sorangium cellulosum in 1977. They display high biological advantages such as strong antifungal action [45][46][47][48][49][50][45,46,47,48,49,50]. The mechanistic studies of these compounds suggested that ambruticins target Hik1 kinase [51][52][51,52] by interacting with fungal osmoregulation. The influence of ambruticin VS3 on soil myxobacteria has recently been studied, and results showed that they are beneficial for the environment by preventing the emergence of antagonistic myxobacterial species. In 2021, Trentadue et al. [53] reported the total synthesis of ambruticin J by utilizing the Corey-Seebach reagent as a key step. For this purpose, dithiane 34 (synthesized from propargyl alcohol) was reacted with epoxide 35 by using the Corey-Seebach reaction to afford compound 36 with a 70% yield, and after a few steps, vinyl iodide 37 was formed. In the following stage, vinyl iodide 37 reacted with pinacol boronic ester 38 through Suzuki coupling, followed by oxidation using Dess–Martin periodinane (DMP) to afford aldehyde 39. The aldehyde 39 was further treated with fragment C via Julia-Kocienski olefination to afford E-olefin 40, and after a few steps, the desired ambruticin J 41 was formed (Scheme 5).
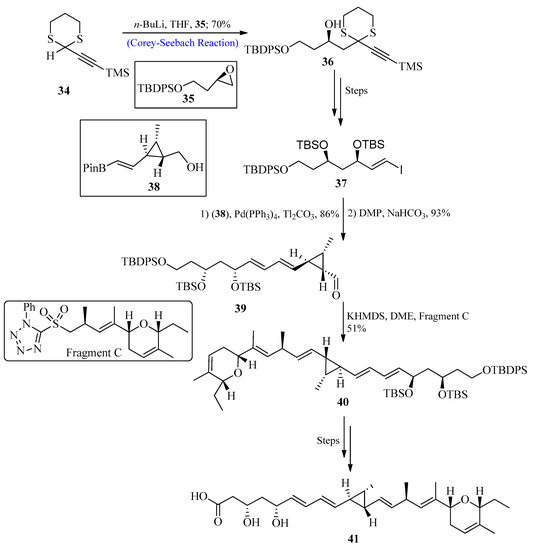
Scheme 5. Synthesis of ambruticin J 41.
4.2. Biakamides
Biakamides are naturally occurring polyketides with significant biological activity [54][55][54,55]. In 2017, Kotoku et al. [56] first isolated biakamides from a marine sponge Petrosaspongia sp. The purpose of this research project was to isolate marine-based anti-cancer drugs. To achieve this, marine-based biakamides were isolated, and the total synthesis of these drugs has also been described by using the Corey-Seebach reaction in one of their key steps. The synthesis was initiated by using substituted penta-diol 42, which was converted into corresponding Weinreb amide 43, followed by reduction with DIBAL to obtain compound 44. Aldehyde 44 was treated with 1,3-propane dithiol in the presence of iodine to afford 1,3-dithiane 45. Compound 45 was then allowed to react with alkyl iodide 46 in the presence of n-BuLi by using the Corey-Seebach reaction followed by TBAF addition and subsequent tetrapropylammonium perruthenate (TPAP) oxidation to furnish aldehyde 47. After a few steps, N-methyle-neamide 48 was synthesized from secondary amine 49, followed by the deprotection of 1,3-dithiane to provide compound 50. The chloromethylene moiety was introduced in the presence of (chloromethyl)triphenyl-phosphonium chloride with E/Z 3:2 by using the Wittig reaction, which resulted in compound 51. In the last step, TFA was used for the deprotection of the amine, followed by a condensation reaction with E-3-methoxy-2-butenoic acid 52 to afford (4R, 6S)-biakamides 53 and 54 (Scheme 6). The antiproliferative activity of biakamides 53 and 54 was also examined against PANC-1 cell culture (glucose deficient conditions), which provided an IC50 value of 0.5 μM.
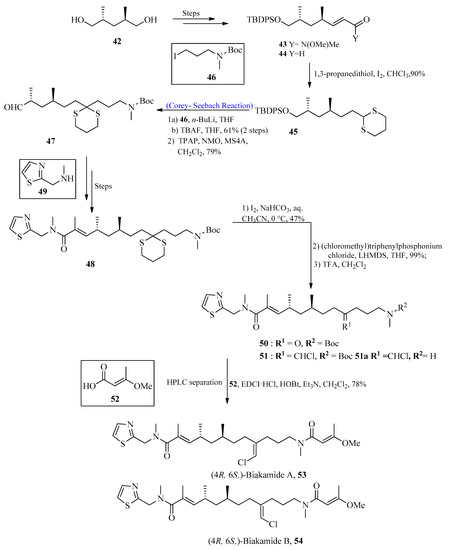
Scheme 6. Total synthesis of biakamides 53 and 54.
5. Photoinitiators
5.1. Bisacyldigermanes
The synthesis of improved photoinitiator molecules for free radical polymerization has been a challenging task. So far, a large number of photoinitiators, such as acyl-phosphine oxides, have been successfully synthesized [57]. Among all types, germanium-based photoinitiators are of great importance due to their non-toxic behavior and excellent bleaching properties [58]. In 2022, Wiesner et al. [59] synthesized bisacyldigermanes 59 by utilizing the Corey-Seebach reaction. The purpose of this synthesis was to introduce double germanium content in order to achieve a higher polymerization rate. For this purpose, 1,2-dichloro-1,1,2,2-tetraethyldi-germane 56 was synthesized over four steps from diethyl dichloro germane 55, followed by lithiation with thioketals 57a–e to afford germane derivatives 58a–e. In the last step, compounds 58a–e were deprotected and oxidized using boron trifluoride etherate and (diacetoxyiodo)benzene (PIDA) to obtain bisacyldigermanes 59a–e in good yields (Scheme 7).
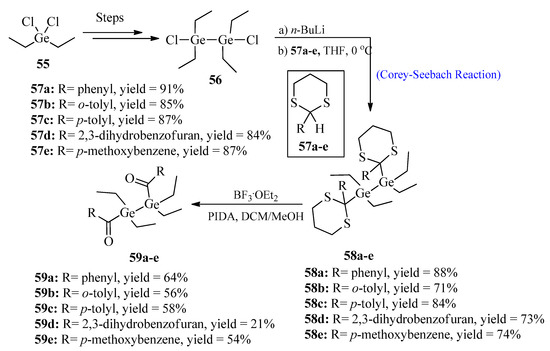
Scheme 7. Synthesis of bisacyldigermanes 59.
5.2. Benzoylgermanium Derivatives
Germanium-based photoinitiators have attained great importance due to their high radical polymerization capacity [60][61][60,61]. In 2009, Moszner et al. [62] synthesized benzoyl germanium derivatives using the Corey-Seebach reaction. These benzoyl germanium derivatives are used in dental cements and composites. In the first step, aromatic 1,3-dithianes 59 were reacted with n-BuLi by using the Corey-Seebach reaction, followed by the reaction with dichlorogermanium compound 60 to afford compound 61a–f. In the last step, compound 61 was dithioketolized in the presence of BF3·OEt2 and Phl(OAc)2 or in the presence of excess iodine and CaCO3 in THF to provide PIs 62a–f (Scheme 8).
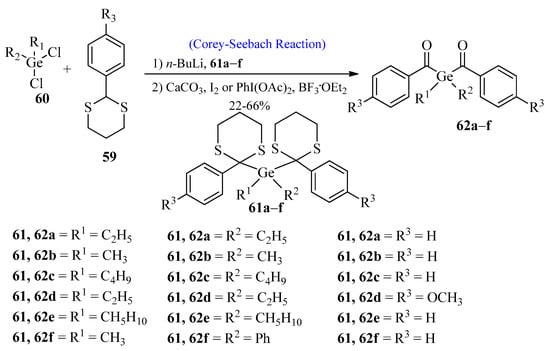
Scheme 8. Synthesis of benzoyl germanium derivatives 61-a-f via Corey-Seebach approach.