Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Qi Sun and Version 2 by Jessie Wu.
Proteins are biomacromolecules widely present in biological processes in vivo with important functions, such as biological catalysis, high-affinity molecule recognition, and activation and/or inhibition of cellular pathways. Polymer-protein systems have excellent characteristics, such as non-toxic, non-irritating, good water solubility and biocompatibility, which makes them very appealing as cancer therapeutics agents.
- polymer
- protein
- cancer therapy
- design
1. Selection of Polymers
The selection of polymers with good properties can maximize the anticancer effect of hybrid polymer–protein systems. The first thing to consider is the size of the polymer. Although larger polymers generally increase the half life, larger sizes make it easier for them to gather, leading to decreased kidney clearance and creating obstacles to metabolism in the body. Therefore, the smallest molecular weight polymer that increases the circulating half-life of the protein to the level required for the intended treatment should be selected. Moreover, the size of the polymer has an effect on biological activity [1][17]. Larger polymers often produce conjugates with lower biological activity, most likely due to non-specific steric hindrance. The ability of polymers to bind to proteins is another factor in the hybrid system. Additionally, the chemical nature of polymers affects the overall architecture of the conjugate. The shape of the polymers or polymer topology affects the properties and functions of the system. Brush-like, hyperbranched or dendritic topologies often have good characteristics compared to their linear counterparts while forming polymer bioconjugates [2][18]. In addition, degradability, half life, immunogenicity and related toxicities of polymers should be considered.
PEG and PEG analog, biomimetic polymer, degradable polymer and stimuli-responsive polymer represent a group of polymers explored in polymer–protein conjugation. Common functional polymers are as follows. Several types of polymers used in hybrid polymer–protein systems are summarized in Table 1.
1.1. Polyethylene Glycol (PEG) and Polyethylene Glycol Analog
1.1. Polyethylene Glycol (PEG) and PEG Analog
PEG, a linear and nonionic polyether diol with the molecular formula HO-(CH2-CH2-O)n-H, is widely used in the modification of hybrid biological polymers, such as proteins and peptides. PEG is one of the most commonly used polymer materials and an excipient in the pharmaceutical industry with many advantages of non-toxic, non-irritating and good water solubility. PEG modification can change the physical and chemical properties of drugs, including conformation, electrostatic binding, hydrophobicity, etc. These physical and chemical changes can increase the retention time of drugs in vivo and prolong absorption time. PEGylation shields antigenic epitopes through steric hindrance, thereby reducing the immunogenicity of the drugs. Additionally, steric hindrance shields the drugs from enzymatic degradation and opsonization with serum proteins. Drug binding affinity to cell receptors can also be affected by PEG modification, thereby reducing nonspecific uptake by the mononuclear phagocyte system and improving the tumor targeting ability of delivery systems [3][4][19,20]. On the other hand, PEG modification can reduce the frequency and incidence of adverse events administered as well as improve drug efficacy and tolerance [5][21]. In addition, PEG can also increase the solubility and stability of proteins which is also beneficial for the production and storage of drugs [6][7][22,23].
In earlier studies, the immunogenicity and antigenicity of PEG were not found, and PEG has been considered a biologically inert material. Since then, PEGylation has evolved dramatically as an important way to improve the pharmacokinetics and pharmacodynamics of protein drugs [8][24]. However, with more and more PEGylated protein drugs entering clinical applications, side effects caused by the administration of PEGylated protein drugs are gradually increasing. For example, PEGinesatide (Takeda, Deerfield, IL, USA) was withdrawn from the market due to severe hypersensitivity reactions with fatal consequences which may be related to PEG [9][10][25,26]. Many reports have claimed that the production of anti-PEG antibodies in patients and the loss of drug efficacy continue to appear [11][12][13][27,28,29].
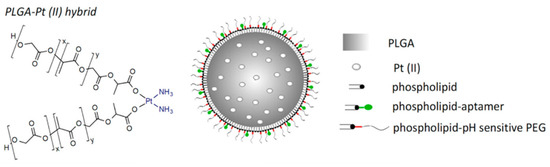
In order to solve these problems, PEG analogs were born on demand. PEG-like brush polymers not only enhance cycling half life but also do not induce an anti-PEG antibody response. For example, poly (polyethylene glycol methyl ether methacrylate)—p (PEGMA) is a methacrylate polymer having a low ethylene glycol side chain that has been used in a variety of protein–polymer conjugates [14][15][30,31]. In addition, poly(N-(2-hydroxypropyl) methacrylamide) (p(HPMA)) is another water-soluble and biocompatible polymer that can be used to increase half life in vivo and decrease associated immunogenicity [16][17][32,33]. Furthermore, zwitterionic poly(carboxybetaine) (PCB) with good biocompatibility and superior hydrophilicity was introduced as a PEG substitute for protein modification. As reported by Li et al. [18][34], the generation of PCB-specific antibodies was minimal and insensitive to increasing protein immunogenicity. A pH-sensitive PEG derivative polymer, poly(lactic-co-glycolic acid) (PLGA), coated with phospholipid-DNA aptamers and an acid-labile hydrazone linkage was designed (Figure 1) [19][35]. The hybrid polymer encapsulating platinum (II) could target a cell-spanning protein overexpressed to more than 90% of late-stage ovarian cancer, mucin 1 (MUC1), thus achieving enhanced cancer therapy. It follows that PEG analogs have a wider application prospect in the field of biomedicine.
1.2. Biomimetic Polymer
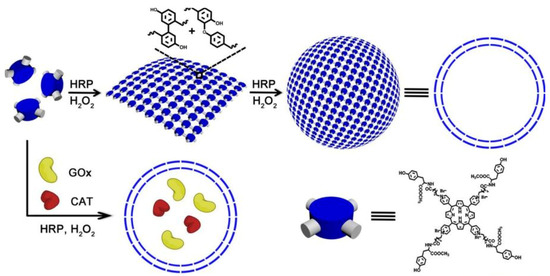
Biomimetic polymer is a kind of polymer that resembles natural polymers in form, appearance and properties with the benefits of synthetic polymers. For example, trehalose can form a special protective film in cells under harsh conditions, such as high temperature, high cold and dry water loss, effectively protecting the structure of biomolecules from being destroyed, thus maintaining the life process and biological characteristics of living organisms [20][21][22][36,37,38]. Therefore, natural disaccharide trehalose polymers can stabilize biological macromolecules under extreme conditions. For example, Diaz-Dussan et al. [23][39] reported a trehalose-based polymer that can be used as a cryoprotectant and three-dimensional (3D) cell scaffold for cell encapsulation and organoid production. Polyelectrolytes (PEs) molecules that carry multiple negative or positive charges may be functionalized by absorbing proteins at the top of the architecture or by inserting one or more kinds of proteins [24][40]. In particular, natural PEs have been extensively investigated for drug delivery because of their readily available, low toxicity, biodegradable and biocompatible properties. Among them, polysaccharides are the most promising materials, such as dextran [25][41], cellulose [26][42], and pectin [27][43], which are naturally derived linear and/or branched polymers widely used as PEG alternatives in the fabrication of therapeutic protein/drug conjugates. Recently, PSA-rFVIII (a novel polysialylated recombinant coagulation factor VIII—BAX 826) has completed a Phase I clinical trial (NCT02716194) in patients with severe hemophilia A and demonstrated long circulation half life with adequate safety and tolerability [28][44]. Another example of biomimetic polymers is heparin-mimicking polymers, such as carboxymethyl benzylamide sulfonate dextrans (CMDBS) [29][45], polysulfonated polymers [30][31][46,47] and sulfated synthetic glycopolymers [32][48]. Covalent dimerization of tyrosine in a horseradish peroxidase (HRP)-catalyzed system was an efficient method for enzyme-catalyzed preparation of polymers. Liu et al. constructed polymer nanocapsules with porphyrin photosensitizers as building blocks based on HRP-catalyzed tyrosine dimerization (Figure 2) [33][49]. The polymer nanocapsules further encapsulated glucose oxidase (GOx) and catalase (CAT) to obtain a biomimetic cascade nanoreactor (GOx/CAT-NC) for starvation and photodynamic cancer therapy which contribute to the advancement of complementary modes of spatiotemporal control of cancer therapy. These biomimetic polymers can stabilize, encapsulate and control release proteins, enhancing therapeutic efficacy and reducing side effects in therapeutic applications.
1.3. Degradable Polymer
Non-degradable polymers can accumulate in the body and may cause potential toxic side effects to the body, so it is necessary to develop degradable polymers for biomedical purposes [34][50]. Cyclic ketene acetal (CKA) is a monomer that produces hydrolyzable backbone ester bonds by a free radical ring-opening reaction [35][51]. CKAs can be copolymerized with vinyl monomers to produce functional aliphatic polyesters which are degradable polymers widely used in medical science [36][37][38][52,53,54]. In addition, there are some natural polymer analogs, such as peptides [39][55] and hydroxyethyl starch (HES) [40][56], which are degradable in vivo. Various proteins have been conjugated to synthetic polypeptides, and although these peptides are stable, they can be degraded in vivo by proteases. Remarkably, several micellar formulations based on degradable poly(ethylene glycol)-b-polypeptide block copolymers (NK105, NC6004, NK911, etc.) have been developed into different phases of clinical trials to treat breast, pancreatic and gastric cancers [41][57]. HES can be degraded by α-amylase in plasma, and its conjugates have been extensively studied for therapeutic applications [42][43][44][58,59,60]. PLGA is a common biodegradable polymer that is usually broken down in the body into LA and GA which is eventually metabolized by the body into carbon dioxide and water. Due to its outstanding biocompatibility, biodegradability and mechanical properties, PLGA has been approved by the FDA and the European Medicines Quality Agency (EMA) as a superior drug carrier. PLGA-based drug delivery systems effectively improve the bioavailability of drugs, reduce adverse effects and reduce the frequency of dosing which may improve patient compliance with medication [45][61]. Biodegradable polymers are both synthetic and natural, and all biodegradable polymers are basically stable and durable in applications. In terms of application, biodegradable polymers are of great significance in medical treatments.
1.4. Stimulus-Responsive Polymer
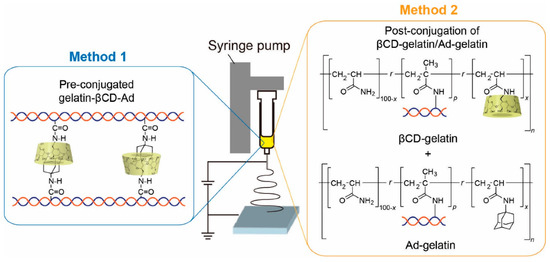
In addition to improving pharmacokinetics, polymers can also impart new properties to proteins to create a stimulus-responsive system. For example, polyacrylamides, such as poly(N-isopropyl acrylamide) (p(NIPAAm)), have a lower critical dissolution temperature which allows polymers to precipitate with increasing temperature [46][62]. There are also conjugates of pH-responsive ionic polymers, such as polyacrylic acid [47][63] and poly (N,N-dimethylaminoethyl methacrylate) (p(DMAEMA)) [48][64]. The use of stimulus-responsive polymers in disease theranostic applications may enable a more controllable and effective process, for example, hyperthermia-mediated cancer treatment [49][65] and acid response nanoprobe in cancer diagnosis [50][66]. Polymer- and/or protein-based nanofibers that promote stable cell adhesion have drawn increasing attention as well-defined models of natural 3D extracellular matrix. Kentaro et al. fabricated two types of stimulus-responsive gelatin-containing supramolecular nanofibers that can be utilized as well-defined, switchable 3D microenvironments for cells. The first type of nanofibers was prepared by coupling the host–guest inclusion complex to gelatin before electrospinning, while the second type of nanofibers was fabricated by coupling gelatin to polyacrylamide functionalized with the host (βCD) and guest (Ad) moieties followed by conjugation in the electrospinning solution (Figure 3) [51][67]. The stimulus responsive was achieved by elasticity switching under physiological conditions by adding/removing soluble guest molecules. In addition to this, there are other polymers that respond to redox agents [52][68], photo irradiation, CO2 [53][69], enzyme [54][70], electric [55][71] and/or magnetic field [56][72], photo radiation [57][73] and so on.
Table 1.
Several types of polymers used in hybrid polymer–protein systems.
| Category | Example | References |
|---|---|---|
| PEG and PEG analog | p(HPMA) | [16][17][32,33] |
| PCB | [18][34] | |
| Poly(lactic-co-glycolic acid) (PLGA) | [19][35] | |
| Biomimetic polymer | Trehalose-based polymer | [23][39] |
| Dextran, Cellulose, Pectin polymer | [25][26][27][41,42,43] | |
| Heparin-mimicking polymer | [29][30][31][32][45,46,47,48] | |
| Horseradish peroxidase (HRP)-catalyzed system | [33][49] | |
| Degradable polymer | CKA based polymer | [35][51] |
| Peptides | [39][55] | |
| HES | [40][56] | |
| PLGA | [45][61] | |
| Stimulus-responsive polymer | p(NIPAAm) | [46][62] |
| polyacrylic acid | [47][63] | |
| p(DMAEMA) | [48][64] | |
| Gelatin-containing supramolecular nanofibers | [51][67] |
These are the main types of polymers above-mentioned. Table 1 shows some examples of different types of polymers.
2. Section of Proteins
In general, polymer–protein coupling faces more spatial and entropy barriers than small-molecule coupling reactions. Therefore, the commonly used coupling methods need to be highly efficient, and the selection of coupling chemistry also needs to consider site selectivity and the availability of residues on proteins. Site-selective coupling is necessary to maintain adequate biological activity. Typically, the conjugated site should be located away from the active site or binding motif to maximize protein activity. Therefore, it is important to select proteins that contain easily modified residues, such as lysine, cysteine and disulfide bonds [58][59][60][74,75,76]. It is worth noting that protein active sites must be predefined, and the binding motif should be away from the conjugation sites so as not to affect protein activity [61][77]. For protein denaturation, protein aggregation during the denaturation process must be strictly avoided. Because it is difficult to disaggregate protein agglomerates once they have precipitated, this reduces yields and hinders the protein purification process. In addition, biocompatibility and biodegradability by proteases, peptide sequence, various functionalities in specific positions and tunable transition also need to be considered for well-defined polymer–protein systems [62][78].
3. Bonding Form of Polymer-Protein Systems
Many active reactive groups are present in both polymers and proteins which allows them to bind in a variety of ways mainly by covalent and non-covalent conjugation.
3.1. Covalent Conjugation
Reactive groups on polymers are electrophiles that react with nucleophilic groups of proteins, such as lysine, cysteine and tyrosine residues, typically at one end of the polymer chain. These common reaction groups include activated esters, carbonates, aldehydes, amine-reactive mercaptide reagents, Michael receptors, disulfide bond exchange, N-terminal coupling, tyrosine conjugation, etc.
Lysine is the most abundant amino acid on the surface of proteins and is often the first choice to attempt nonselective conjugation. Although lysine is not commonly used for site-selective conjugation, it is still very effective at loading conjugation due to its amino group. Lysine has a positively charged ε-amino group and the group requires a neutral to basic pH to have sufficient nucleophilicity (pKa~10.5). The pKa (6–8) of N-terminal amine is significantly lower than that of lysine, selective N-terminal modification is usually pH-dependent on the N-terminal α-amino group [63][79]. Common lysine coupling reactions involve the formation of stable amide bonds using a payload of active esters, e.g., o-succinimide reagents N-hydroxysuccinimide (NHS) [64][65][80,81]. Lysine reacting with maleimide [66][82] or halogenated acetamide [67][83] and coupling by isothiocyanate [68][84] to form stable thiourea bonds are also conjugation methods.
The high nucleophilicity of cysteine makes it easy to modify, but free cysteine is rare and is usually located in hydrophobic pockets. On the one hand, disulfide formation is a kind of cysteine modification. Cysteine is present in the form of disulfide bonds in antibodies and only free or reduced sulfhydryl groups (–SH) are available for reaction with thiol-reactive compounds, so reducing free sulfhydryl groups by using dithiothreitol, thioglycerol, or other sulfhydryl reducing reagents is necessary for the next step of the reaction [69][70][71][85,86,87]. On the other hand, alkylation formation by using carbonyl compounds to form thioethers can achieve cysteine modification [72][88]. Cysteine may also be modified by maleimide [73][89] or vinyl sulfone [74][90] functionalized groups by Michael addition at neutral pH.
Disulfide bonds are present in most proteins, playing an important role in maintaining spatial and resulting biological activity. They can be used as residue-specific binding sites. Tian et al. [75][91] prepared a multifunctional nanocarrier by anchoring transferrin to hollow mesoporous silica nanoparticles through disulfide bonds which can be resolved in the presence of glutathione, achieving redox response.
Although tyrosine has a low utilization rate, most proteins contain tyrosine residues with a natural abundance of about 3.2% [76][92], and it can be modified through nitrating agents. Several coupling methods, such as diazotization coupling [77][93] and 4-phenyl-3H-1,2,4-triazoline-3,5(4H)-dione (PTAD) coupling [78][94], have been developed. In addition, histidine [79][95], glutamine [80][96] and aldehyde [81][97] tag modifications are also considered important modification sites.
On a technical level, grafting synthesis of polymers to proteins can be achieved by using these three synthetic strategies: grafting to, grafting from and grafting through [82][98]. In brief, grafting to is the coupling of a pre-synthesized polymer with a biomolecule, where a pre-formed reactive polymer is conjugated to a protein using a backbone chain with functional groups that are randomly distributed along the chain. Graft copolymer formation originates from the coupling reaction between the functional backbone and reactive branch end groups. Grafting from is a technique involving monomers that are polymerized using an initiation reaction on the membrane surface, where the polymer chain is grown from a protein macroinitiator. The grafting from process involves in situ growth of a polymer from a biomolecule or alternatively synthesis of a biomacromolecule using a preformed polymer as the initiator [83][99]. Both strategies can be followed using protein-reactive RAFT agents [84][85][100,101]. For example, a water-soluble RAFT agent was conjugated to a model protein, bovine serum albumin (BSA), via its free thiol group at Cys-34 residue [86][102]. In addition, protein-reactive RAFT chain transfer agents contain either NHS or pentafluorophenyl ester moiety that can bind to lysine residues, and alternatively maleimide or pyridyl disulfide moiety for binding to cysteine residues [87][103]. The use of these two techniques is more frequent than grafting through, also known as the macromonomer method, which is one of the simpler ways to synthesize a graft polymer with well-defined side chains. Grafting through can polymerize biomolecule-containing monomers yielding bioconjugates with multiple biofunctional groups along the polymer backbone. For example, Dan et al. reported a well-controlled synthesis of molecular brushes through the grafting through method by aqueous ATRP, in which oligo- and poly(2-oxazoline) were used as macromonomers for polymerization [88][104].
3.2. Non-Covalent Conjugation
Non-covalent conjugation methods mainly include electrostatic adsorption, hydrogen bonding, hydrophobic, host−guest interaction [89][90][105,106], etc. Electrostatic adsorption is a common form of connection between proteins and polymers. The charged region of proteins can greatly facilitate proteins’ interaction with other molecules or surfaces. Due to their hydrophilic nature, charged amino acids tend to be located on the outside of proteins, where they can interact with surfaces. In terms of surface chemistry, protein adsorption is a key phenomenon that describes the aggregation of these molecules outside the material. The ability of a protein to remain attached to a surface depends largely on material properties, such as surface energy, texture and relative charge distribution. The size and spatial structure of proteins also affect adsorption capacity [91][107]. Due to the greater number of contact points between amino acids and the surface of the material, larger proteins are more likely to adsorb and remain attached to the surface. The temperature and size of side chains in polymers also affect the strength of the adsorption force. Tan et al. [92][108] found that the affinity of the hIgG2 monoclonal antibody (mAb) with the negatively charged copolymer-modified surface was higher for adsorptions at 50 °C compared with 20 °C. Because at this higher temperature (50 °C), hydrophobic and electrostatic interactions were enhanced. In addition, the size of the side chains in the copolymer structure also influences the electrostatic interactions between the copolymer-modified surface and protein molecules. In addition, changes in electrostatic force will facilitate the adsorption/desorption process for controlled-release systems. Kwon et al. [93][109] reported the electrostatic interaction between human lysozyme and negatively charged albumin-heparin microspheres was affected by ion exchange. While changing the charge in the solution, an adsorption/desorption process was promoted, achieving a controlled release of positively charged polypeptides and proteins.
Hydrogen bonds and hydrophobic interactions are also widely found to hold proteins and polymers together. Hydrogen bonds can be defined as bonds formed between highly electronegative atoms, such as fluorine, oxygen, nitrogen, sometimes chlorine, etc. and hydrogen atoms. In contrast, hydrophobic interaction is a multifaceted phenomenon [94][110]. Briefly, non-polar molecules or groups of molecules attract each other and gather in the water environment, and the original water molecules near the non-polar groups are crowded out, resulting in an increase in the entropy of the surrounding water molecules. Hydrogen bonds and hydrophobic interactions often play a key role in the formation and self-assembly of polymer–protein hybrid systems. For example, polyamidoamine (PAMAM-G4) conjugated with serum albumin via hydrophobic and hydrogen-bonding interaction, achieving high protein loading efficacy (45–55%) [95][111]. Cai et al. [96][112] reported that hydrophobic interactions induced poly(N-isopropylacrylamide) (PNIPAM) and bovine serum albumin (BSA) to form polymer–protein hybrid self-assembly complexes in aqueous solution, while enhancing BSA stability (retaining over 90% of its activity). In addition, host–guest interaction is also commonly used in the formation of polymer–protein self-assembly complexes. For instance, a water-soluble pillar [97][5] arene and a lysine derivative were used for host–guest complexation, and the obtained complex was employed in the formation of drug-loaded vesicles for controllable drug release [98][113].
The main bonding forms of polymer–protein systems are summarized in Table 2.
Table 2.
Bonding forms of polymer-protein systems.
| Category | Example | References |
|---|---|---|
| Covalent conjugation | Amide bond | [64][65][80,81] |
| Thiourea bond | [68][84] | |
| Thioether bond | [73][89] | |
| Disulfide bond | [75][91] | |
| Non-covalent conjugation | Electrostatic adsorption | [92][93][108,109] |
| Hydrogen bond | [95][111] | |
| Hydrophobic interaction | [96][112] | |
| Host–guest interaction | [98][113] |
4. Consideration in the Design Process
For cancer treatment systems, proteins often play therapeutic or adjuvant therapeutic roles alone or in synergy with polymers. During the design process of anti-cancer systems, reasonable polymer–protein binding and release methods must be considered according to anti-cancer treatment strategies. A representation of protein loading and release methods is shown in Figure 4.
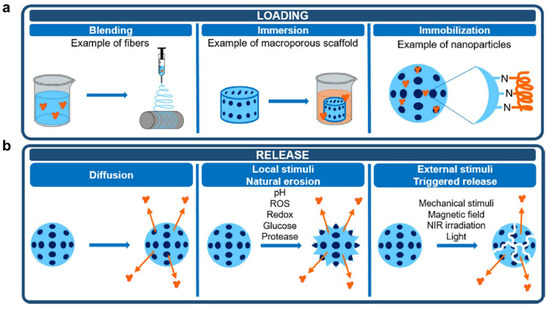
Commonly protein loading steps include (Figure 4a): (1) Polymers blend with protein in a solution at an appropriate temperature. In this process, the choice of temperature and solvent is important to ensure that proteins and polymers activity is not affected. (2) During immersion, proteins absorb to the surface and/or interior of polymers. This process can be assisted by stirring, so that proteins and polymers can contact more evenly. (3) The immobilization process allows proteins to bind to polymers by covalent bonds or non-covalent bonds mentioned above, forming ultimate systems. The entire loading process should ensure that protein activity is not affected by external conditions. Meanwhile, reaction solvents, temperature, pH, stirring speed, etc. can be controlled to prepare a complex system with suitable particle size and protein loading for subsequent treatment. In addition, the protein’s binding to the polymer is also crucial. The desirable linker or spacer must be selected to be adequately stable during circulation, protect the drug from premature metabolism and facilitate the release of drugs enzymatically or hydrolytically cleavage. In addition, other anti-cancer drugs or adjuvants can also be loaded at the same time during the protein loading process to achieve one-pot preparation. In another embodiment, proteins are added during polymerization to form polymer–protein complexes or polymer-coated protein systems, e.g., liposomes loaded with proteins.
In a polymer–protein system, proteins often need to be released for their function to develop. The release methods include (Figure 4b): (1) Proteins self-diffuse through the matrix, leading to desorption of the systems. This process is slow and difficult to release proteins completely. (2) Local stimuli, such as pH, redox, ROS, glucose and protease can contribute to the natural erosion of the system, promoting the release of proteins and other drugs. Especially for cancer therapy, the tumor microenvironment is characterized by low pH and high H2O2 levels which is ideal for responsive drugs to play their roles. (3) Some external stimuli, such as mechanical stimuli, visible or near-infrared (NIR) irradiation, magnetic field, etc. can induce the degradation of the system and reduce the binding forces between polymers and proteins, boosting the controlled release of proteins.
In order to obtain an ideal system, other details are also worth considering, such as biocompatibility of materials, the proteins’ density on the polymers, distance from the contact surface, the release rate and manner of proteins, the possible hazards of responsiveness, etc.