Phosphatidylinositol 3-kinases (PI3Ks) are a family of enzymes responsible for phosphorylating the 3-hydroxyl group of the inositol ring of phosphatidylinositol. PI3Ks are subdivided into three classes, class I, II, and III based on their structures, functions, and tissue distribution. PI3Ks are engaged in a range of cellular functions such as cell growth, proliferation differentiation, migration, survival, cell death, and intracellular trafficking. Many of these functions are related to the activity of class I PI3Ks family. Class I PI3Ks family generates phosphatidylinositol 3,4,5-triphosphate (PI(3,4,5,)P3), which activates an assorted number of downstream molecules in the PI3K signaling pathway that are necessary for normal cellular growth and development. Elevated PI3K activity is considered as the hallmark of cancer, which is promoted by diverse oncogenes and growth factor receptors causing enhanced PI3K signaling. Several PI3K pathway inhibitors have been developed including pan-PI3K inhibitors and isoform-specific PI3K inhibitors. Some of the PI3K inhibitors have been approved for the treatment of various cancer types. Furthermore, to overcome drug resistance, improve efficacy, enhance synergy, and reduce toxicity, several different therapeutic approaches are being developed and some are in clinical trials.
- Phosphatidylinositol 3-kinase
- pan-PI3K inhibitors
- Signaling
- Oncogenes
1. Class I Phospatidylinositol 3-Kinases
Class I PI3Ks are involved in diverse pathways activated by an array of signals including growth factors, mitogenic factors, and hormones [1][2]. Class I PI3Ks are different in their tissue distribution and functions. While PI3Kα and PI3Kβ isoforms are ubiquitously expressed, PI3Kγ and PI3Kδ isoforms are enriched in hematopoietic cells such as leukocytes [3]. Class I PI3Ks are further subdivided into class IA (PI3Kα, PI3Kβ, and PI3Kδ) and class IB (PI3Kγ) and are heterodimers composed of a regulatory subunit (p85 or p101 family) and a catalytic subunit (p110α, p110β, p110γ or p110δ). All four class I catalytic subunits, p110α, p110β, p110γ, and p110δ are encoded by PIK3CA, PIK3CB, PIK3CG, and PIK3CD genes, respectively. The p85 regulatory subunit consists of p85α (p85α, p55α, or p50α splice variants), p85β and p85γ and is encoded by PIK3R1, PIk34R2, and PIKR3 genes, respectively. The catalytic subunits p110α, p110β, and p110δ associate with regulatory subunit p85 to form class IA PI3Kα, PI3Kβ, and PI3Kδ. The catalytic subunit p110γ and regulatory subunit p101 together form class IB PI3Kγ. All four class I PI3Ks catalyze the conversion of phosphatidylinositol 4,5-biphosphate [PI(4,5)P2] to phosphatidylinositol 3,4,5-triphosphate [PI(3,4,5,)P3]. PI(3,4,5,)P3 (PIP3) is a second messenger involved in the activation of a number of downstream molecules of the PI3K signaling pathway [1][2][4].
PI3Ks transduce a host of cellular signals and regulate a wide range of essential cellular processes and physiological functions, including cell growth, proliferation, differentiation, survival, migration, death, and metabolism [1][2][5]. Obviously, the PI3K pathway is highly regulated by multiple mechanisms, often involving crosstalk with other signaling pathways in various physiological and pathophysiological situations. Considering that the PI3K signaling is fundamental to normal cellular processes and physiological functions, glitches in the regulation of the PI3K pathway can lead to the pathogenesis of diseases. Overwhelming information indicates aberrant activation of the PI3K signaling is strongly linked to human cancers [6][7][8][9]. Non-cancerous neurological disorders [10][11], cardiovascular diseases [12][13][14], diabetes [15], immune disorders [3], and CLOVES syndrome characterized by localized tissue outgrowths [16] also share a common etiology with oncogenesis.
Abnormal mutational activation or amplification of the PIK3CA gene encoding p110α catalytic subunit of PI3Kα is the commonly observed event that is associated with a dysfunctional PI3K signaling cascade in different human solid and hematological malignancies [6][7][8][9]. The PI3K pathway is also dysregulated through other mechanisms including loss or inactivation of phosphatase and tensin homolog on chromosome 10 (PTEN), a tumor suppressor, and upregulation of tyrosine kinase growth factor receptors (RTKs) and oncogenes such as ras [8][9][17][18][19][20]. Recently, activation of PIK3CB gene encoding catalytic subunit p110β of PI3Kβ appears to be involved in the development of prostate and breast cancer [21][22]. Studies also indicate p85 regulatory isoforms also contribute to tumorigenesis by different mechanisms. Alterations in PIK3R1 and PIK3R2 genes encoding p85α and p85β, respectively, are reported to be associated with tumorigenic activity [23][24][25]. Due to these alterations in class I PI3Ks, the PI3K pathway is overactivated in many malignancies leading to activation of Akt/PIKB and downstream effector molecules promoting competitive growth and survival advantage, metastatic ability, and resistance to drug therapy. More than 50% of breast, prostate, ovarian, glioblastoma multiforme, uterine, and lung cancer cases are linked to the deregulation of the PI3K pathway [26][27][28].
2. Activation of Phosphatidylinositol 3-Kinase Signaling
In the absence of activating signals, the kinase activity of the p110 catalytic subunit of class IA PI3K is inhibited by its dimerization with the regulatory p85 subunit. Upon RTK or G-protein coupled receptor (GPCR) activation in response to extracellular signals, class IA p110/p85 heterodimer is recruited to the plasma membrane, where p85 separates from p110 catalytic subunit activating p110 kinase activity [29][30]. Activated p110 phosphorylates phosphatidylinositol 4,5-biphosphate (PIP2) to produce lipid second messenger phosphatidylinositol 3,4,5-triphosphate (PIP3). The PIP3 then binds and recruits a subset of pleckstrin-homology (PH), FYVE, Phox (PX), C1, C2, or other lipid domains of downstream targets to the cell surface membrane. Among these downstream targets, Akt and PDKI are the PH domain-containing proteins that are crucial for activating cell growth and survival pathways. Akt, one of the PI3K effector residing in the cytoplasm in an inactive form, translocates to the plasma membrane upon receptor activation of PI3K and binds to PIP3. Similarly, PDK1, also containing PH domain, translocates to the plasma membrane and binds to PIP3 upon PI3K activation. Upon translocation to the cell surface membrane, PDK1 partially activates Akt by phosphorylating Thr307. Full activation of Akt is achieved through phosphorylation of Ser473 by mTOR complex 2 (mTORC2), DNA-dependent kinase, or PDK2. Akt acts downstream to PI3K controlling multiple pathways that regulate cell proliferation, growth, survival, death, and metabolism through its downstream targets [1][2][5][31]. The other essential downstream target of PI3K is mTOR, which regulates protein synthesis essential for cell growth, proliferation, angiogenesis, and other cellular activities. It acts both downstream and upstream of Akt and it is active in two different multiprotein complexes, the target of rapamycin complex 1 (TORC1) and 2 (TORC2). The central role of PIP3 formed by the class I PI3Ks in all these cellular processes is negatively regulated by PTEN. PTEN downregulates class I PI3K activated pathways and PIP3 levels by converting PIP3 to PIP2 (Figure 1) [1][5][2][32][31][33].
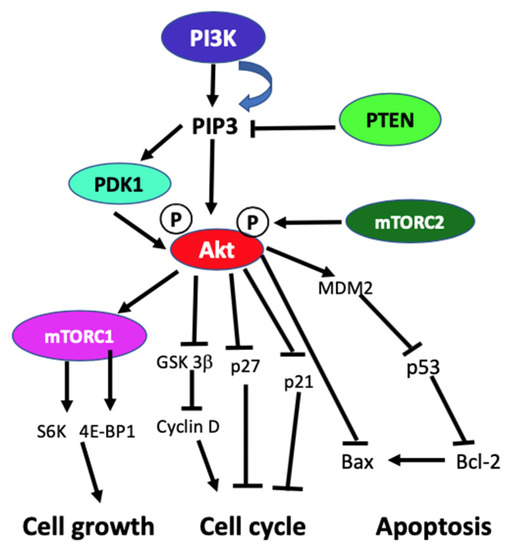
Figure 1. Schematic diagram of generic PI3K signaling displaying the signaling pathways of proteins leading to different cellular effects. In the figure, arrows indicate activation, and blunt end lines indicate inhibition.
3. Phosphatidylinositol 3-Kinase Inhibitors
PI3KIs are subdivided into pan-PI3K inhibitors, isoform-specific PI3K inhibitors, dual PI3K/mTOR inhibitors, mTOR, and Akt inhibitors [32][34][35]. Many of the PI3KIs are being used in preclinical and clinical trials to treat cancers. The pan-PI3KIs such as pictilisib (Taselisib, GDC-0941), apitolisib (GDC-0980), and buparlisb (BKM120) that target all class I isoforms exhibit similar efficacy. Since PIK3CA is frequently mutated in solid tumors, isoform-specific PI3KIs such as alpelisib (BYL719) and taselisib (GDC0032) that selectively target the PIK3CA protein product p100α are under preclinical and clinical testing. On the other hand, B cell signaling is upregulated in chronic lymphocytic leukemia through the hyperactivation of the PI3K pathway due to increased expression of PIK3CD protein product p110δ in hematopoietic cells and lymphoid tissue. Idelalisib, a specific inhibitor for p110δ, is the first PI3KI approved for the treatment of relapsed chronic lymphocytic leukemia in combination with CD-20 antibody rituximab [28]. However, clinical trials with early versions of PI3KIs used as single-target drugs in monotherapy have limited efficacy potentially due to compensatory or concurrent activation of other survival and growth-related signaling pathways [32][34]. Different and alternative strategies are being developed to block tumor growth and progression to overcome the limitations of a single target approach. Among these, the latest polypharmacology approach that is designed to overcome the limitations of single-target therapy appears to have a superior therapeutic effect along with reduced adverse reactions and diminished potential drug resistance. Specifically, the epigenetic polypharmacology approach which uses a dual-acting HDAC and PI3K inhibition by incorporating HDAC inhibitor functionality into a PI3K inhibitor pharmacophore is capable of potent anticancer activity by disrupting the oncogenic signaling network through simultaneous inhibition of HDACs and PI3K in cancer cells [36][37].
References
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanism of isoform-specific PI3K signaling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341.
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signaling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203.
- Okkenhaug, K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu. Rev. Immunol. 2013, 31, 675–704.
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657.
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619.
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garrqway, L.A.; Golub, T.R.; Meyerson, M.L.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501.
- Gyori, D.; Chessa, T.; Hawkins, P.T.; Stephens, L.R. Class (I) phosphoinositide 3-kinases in the tumor microenvironment. Cancers 2017, 9, 24.
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24.
- Janku, F.; Yap, T.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making head way? Nat. Rev. Clin. Oncol. 2018, 15, 273–291.
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox Res. 2019, 35, 775–795.
- Gross, C.; Bassell, G.J. Neuron-specific regulation of class I PI3K catalytic subunits and their dysfunction in brain disorders. Front. Mol. Neurosci. 2014, 7, 12.
- Oudit, G.Y.; Sun, H.; Kerfant, B.-G.; Crackower, M.A.; Penninger, J.M.; Backk, P.H. The role of phoisphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J. Mol. Cell. Cardiol. 2004, 37, 449–471.
- Eisenreich, A.; Rauch, U. PI3K inhibitors in cardiovascular disease. Cardiovasc. Ther. 2011, 29, 29–36.
- Durrant, T.N.; Hers, I. PI3K inhibitors in thrombosis and cardiovascular disease. Clin. Trans. Med. 2020, 9, 8.
- Maffei, A.; Lembo, G.; Carnevale, D. PI3Kinases in diabetes mellitus and its related complications. Int. J. Mol. Sci. 2018, 19, 4098.
- Kurek, K.C.; Luks, V.L.; Ayturk, U.M.; Alomari, A.I.; Fishman, S.J.; Spencer, S.A.; Mulliken, J.B.; Bowen, M.E.; Yamamoto, G.L.; Kozakewich, H.P.W.; et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am. J. Hum. Genet. 2012, 90, 1108–1115.
- Parsons, R. Human cancer, PTEN, and the PI-3 kinase pathway. Semin. Cell Dev. Biol. 2004, 15, 171–176.
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296.
- Aziz, S.A.; Davies, M.; Pick, E.; Zito, C.; Jilaveanu, L.; Camp, R.L.; Rimm, D.L.; Kluger, Y.; Kluger, H.M. Phosphatidylinositol 3-kinase as a therapeutic target in melanoma. Clin. Cancer Res. 2009, 15, 3029–3036.
- Zhou, B.P.; Hu, M.C.; Miller, S.A.; Yu, Z.; Xia, W.; Lin, S.Y.; Hung, M.C. Her-2/neu blocks tumor necrosis factor-induced apoptosis via the Akt/NF-kappaB pathway. J. Biol. Chem. 2000, 275, 8027–8031.
- Hill, K.; Kalifa, S.; Das, J.R.; Bhatti, T.; Gay, M.; Williams, D.; Taliferro-Smith, L.; De Marzo, A.M. The role of PI 3-kinase p100beta in Akt signaling, cell survival, and proliferation in human prostate cancer cells. Prostate 2010, 70, 755–764.
- Dbouk, H.; Khalil, B.D.; Wu, H.; Shymanets, A.; Nurnberg, B.; Backer, J.M. Characterization of tumor-associated activating mutation of the p110β PI 3-K. PLoS ONE 2013, 8, e63833.
- Urick, M.E.; Rudd, M.L.; Godwin, A.K.; Sgroi, D.; Merino, M.; Bell, D.W. PIK3R1 (p85alpha) is somatically mutated at high frequency in primary endometrial cancer. Cancer Res. 2011, 71, 4061–4067.
- Wu, H.; Shekar, S.C.; Flinn, R.J.; El-Sibai, M.; Jaiswal, B.S.; Sen, K.L.; Janakiraman, V.; Seshagiri, S.; Gerfer, G.J.; Girvin, M.E.; et al. Regulation of class IA PI3-kinases: C2 domain- iSH2 domain contacts inhibit p85/p110alpha and are disrupted in oncogenic p85 mutants. Proc. Natl. Acad. Sci. USA 2009, 106, 20258–20263.
- Cortes, I.; Sanchez-Ruiz, J.; Zuluaga, S.; Calvanese, V.; Marques, M. p85beta phosphoinositide 3-kinase subunit regulates tumor progression. Proc. Natl. Acad. Sci. USA 2012, 109, 11318–11323.
- Engelman, J.A. Targeting PI3K signaling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562.
- Spangle, J.M.; Roberts, T.M.; Zhao, J.J. The emerging role of PI3K/Akt-mediated regulation in cancer. Biochem. Biophys. Acta 2017, 1868, 123–1131.
- Furman, R.R.; Sharman, J.P.; Coutre, S.E.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.; et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2014, 370, 997–1007.
- Ramesh, L.E.; Chen, C.S.; Cantley, L.C. Phosphatidylinositol (3,4,5)P3 interacts with SH2 domains and modulates PI 3-kinase association with tyrosine-phosphorylated proteins. Cell 1995, 83, 821–830.
- Yu, J.; Wjasow, C.; Backer, J.M. Regulation of p85/p110alpha phosphatidylinositol 3-kinase. Distinct roles for the n-terminal and c-terminal SH2 domains. J. Biol. Chem. 1998, 273, 30199–30203.
- Manning, B.D.; Toker, A. Akt/PKB signaling: Navigating the network. Cell 2017, 169, 381–405.
- McCubery, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L. Franklin, Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: How mutations can result in therapy resistance and how to overcome resistance. Oncotarget 2012, 3, 1068–1111.
- Milella, M.; Falcone, I.; Conciatori, F.; Incani, U.C.; Curatolo, A.D.; Inzerilli, N.; Nuzzo, C.M.A.; Vaccaro, N.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple functions in human malignant tumors. Front. Oncol. 2015, 5, 24.
- Wee, S.; Jagani, Z.; Xiang, K.X.; Loo, A. PI3k pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009, 69, 4286–4293.
- Ong, P.S.; Wang, L.Z.; Dai, X.; Tseng, S.H.; Loo, S.J.; Sethi, G. Judicious toggling of mTOR activity to combat insulin resistance and cancer: Current evidence and perspectives. Front. Pharmacol. 2016, 7, 395.
- deLera, A.R.; Ganesan, A. Epigenetic polypharmacology: From combination therapy to multitarget drugs. Clin. Epigenet. 2016, 8, 1–21.
- Tomaselli, D.; Lucidi, A.; Rotili, D.; Mai, A. Epigenetic polypharmacology: A new frontier for epi-drug discovery. Med. Res. Rev. 2020, 40, 190–244.