Pituitary neuroendocrine tumors (PitNETs), the third most common intracranial tumor, are mostly benign. However, some of them may display a more aggressive behavior, invading into the surrounding structures. While they may rarely metastasize, they may resist different treatment modalities. Several major advances in molecular biology in the past few years led to the discovery of the possible mechanisms involved in pituitary tumorigenesis with a possible therapeutic implication. The mutations in the different proteins involved in the Gsa/protein kinase A/c AMP signaling pathway are well-known and are responsible for many PitNETS, such as somatotropinomas and, in the context of syndromes, as the McCune–Albright syndrome, Carney complex, familiar isolated pituitary adenoma (FIPA), and X-linked acrogigantism (XLAG). The other pathways involved are the MAPK/ERK, PI3K/Akt, Wnt, and the most recently studied HIPPO pathways. Moreover, the mutations in several other tumor suppressor genes, such as menin and CDKN1B, are responsible for the MEN1 and MEN4 syndromes and succinate dehydrogenase (SDHx) in the context of the 3PAs syndrome.
- pituitary neuroendocrine tumors (PitNETs)
- pituitary adenoma
- pituitary tumorigenesis
- pituitary pathogenesis
- genetic alterations
1. Introduction
2. Cell Signaling Pathways in Pituitary Tumorigenesis
2.1. Gsa/Protein Kinase A/c AMP Signaling Pathway
The G-protein-coupled receptor (GPCR) signaling pathway represents one of the most crucial signaling cascades in development, normal physiology, and disease, and c-AMP, the major second messenger affected by this activation, is the first one described in [20][19]. GPCRs transmit extracellular signals mainly through heterotrimeric G proteins. Their molecule consists of three main subunits, Ga, Gβ, and Gγ (Figure 1). The Ga subunit is the one that defines the nature of each G-protein involved in the hormone action. It can be stimulating (Gs) (activating adenyl cyclase (AC) and increasing the cytosolic c AMP levels) and inhibitory (Gi/o/z) (inhibiting adenylyl cyclase, decreasing the intracellular cAMP levels, and regulating Ca and K as well). Moreover, it can act through the stimulation of phospholipase C Gaq(Gq/11) [21][20]. Nevertheless, these signaling pathways commonly overlap [22,23][21][22].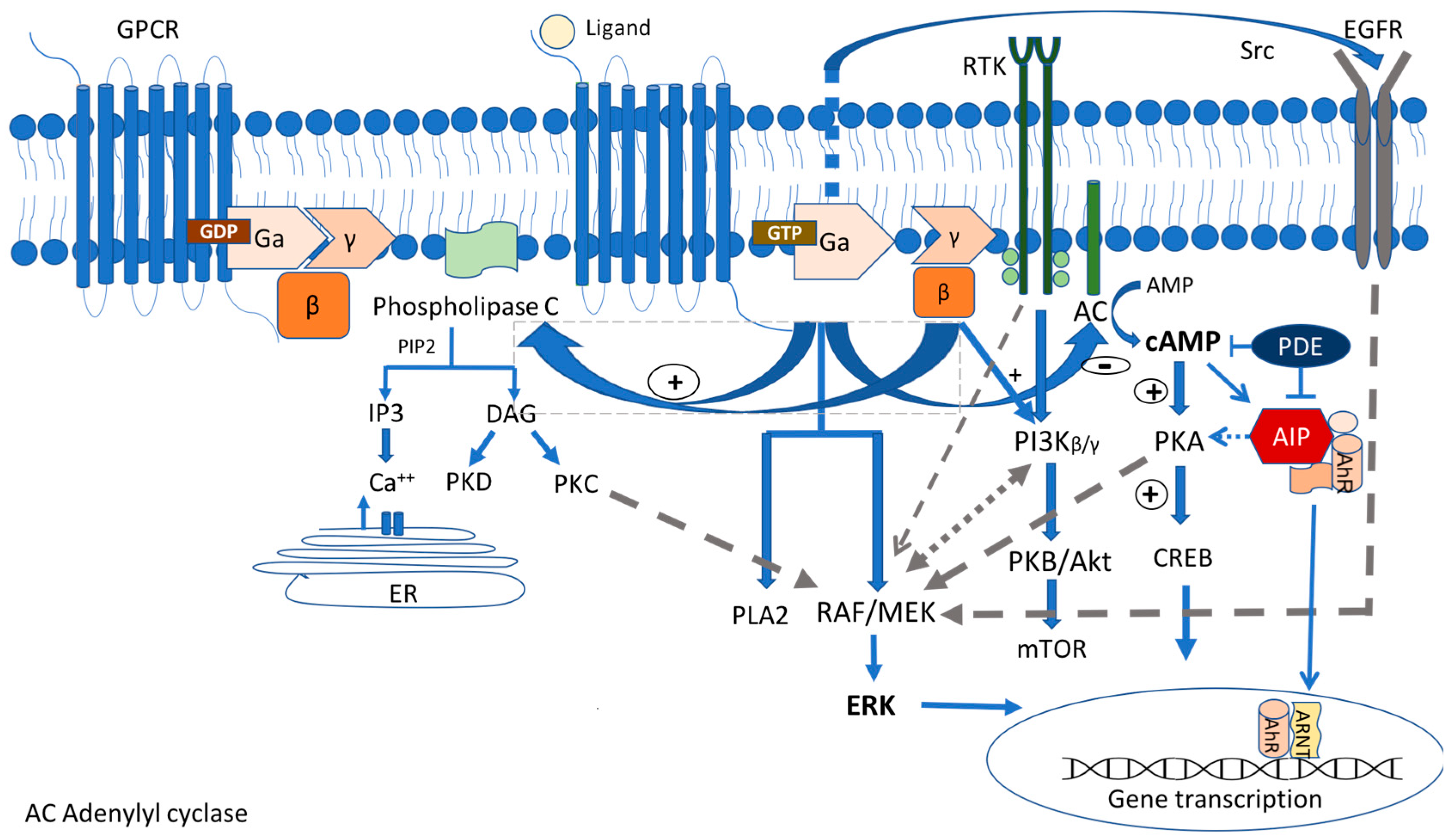
2.1.1. GNAS Mutations
2.1.2. Protein Kinase A Mutations
The Carney complex (CNC) is a rare genetic syndrome inherited in an autosomal dominant manner. In some cases, it occurs sporadically due to de novo mutations. It is characterized by the presence of multiple cardiac and extracardiac myxomas, spotty skin pigmentation, schwannomas and endocrine tumors, such as GH-secreting PitNETs, corticotroph tumors, and ACTH-independent Cushing syndrome known as primary pigmented nodular adrenocortical disease (PPNAD), and thyroid and gonadal tumors [51,52][37][38]. The mutations in the two loci were identified as 17q22-24 and 2p16, which contain the genes that are potentially responsible for the disease (initially known as CNC1 and CNC2). Up to 75% of patients may have an asymptomatic elevation of GH, insulin growth factor I (IGF-1), and prolactin. Approx. 65% of them may exhibit somatomammotrophic hyperplasia (SH), while only 10–12% of them carry PitNETs [55[39][40],56], resulting in gigantism or acromegaly depending on the age of the presentation. Apart from acromegaly, there are some rare reports of lactotroph adenomas [57][41] as well as corticotroph tumors [58,59][42][43], although the ACTH-independent Cushing syndrome prevails in patients with the Carney complex.2.1.3. AIP
Familial isolated pituitary adenomas (FIPA), firstly recognized in 1999, are characterized by the presence of PitNETs in two or more members of the same family without other clinical features found in the context of a syndrome, such as in MEN1, MEN4, Carney complex, or succinate dehydrogenase (SDHx)-related tumors [62,63][44][45]. Approx. 20% of a FIPA harbor germline loss-of-function mutation in the aryl hydrocarbon receptor-interacting protein (AIP) gene map on the chromosome 11q13.3 locus [64][46]. However AIP mutations have been recognized in sporadic PitNETs, particularly those that occur during childhood/adolescence and early adulthood, probably explained by the incomplete penetrance of the disease (approx. 30%) [65,66][47][48]. AIP patients usually have macrotumors, with the first onset of symptoms occurring in childhood/adolescence in about 50% of patients. The AIP is a co-chaperone protein that is expressed in many tissues and has a tumor suppressor function. It is able to bind to different partners using three antiparallel tetratricopeptide a-helix motifs (TPR domains), resulting in multiple protein–protein interactions [68][49]. The loss-of-function AIP mutations lead to a disruption of these interactions, probably contributing to pituitary tumorigenesis [64][46]. One of the most critical interactions is with PDEs, particularly PDE4A5, leading to decreased enzymatic activity and, therefore, negatively regulating the cAMP pathway in the pituitary gland [68,69][49][50]. However, the impact of the loss of this interaction in the context of an AIP mutation is still not completely understood and multiple post-receptor mechanisms and other signaling pathways are involved in pituitary tumorigenesis [70][51]. In addition to the involvement in the c AMP pathway, the AIP exerts its effects by binding and stabilizing the aryl hydrocarbon receptor (AhR), which is best known for mediating the effects of environmental toxins, such as dioxin, the so-called “dioxin receptor”. The AhR is a member of the basic helix-loop-helix/Per-Arnt-Sim (bHLH/PAS) family of transcription factors that regulates the response to halogenated hydrocarbons. It is involved in different cell responses and the regulation of the cell cycle and differentiation. In the cytoplasm, it is stabilized by forming a multimeric AIP/AhR/Hsp90/p23 complex [68][49], avoiding the AhR degradation. Upon ligand binding, the AhR disengages and translocates to the nucleus, where it binds to the aryl hydrocarbon nuclear translocator (ARNT). AIP-mutated pituitary tumors have a broad clinical spectrum. GH-secreting PitNETs usually have an aggressive profile, higher levels of GH and IGF1, and show a resistance to the treatment using first-generation SSAs-octreotide and lanreotide [65][47]. Thus, a low AIP tumor expression is an indicator of tumor aggressiveness and treatment resistance [77][52]. Chahal et al. suggested that octreotide may increase the expression of the tumor suppressor gene ZAC1, and the loss of expression of ZAC1 occurring in AIP-mutated adenomas results in an SSA resistance [78][53]. Dutta et al. reported a four-year-old child with an AIP pituitary macrotumor, which required multimodal treatment with surgery, long-acting octreotide, radiotherapy, temozolomide, bevacizumab, and pegvisomant to be controlled [79][54]. However, not all AIP-mutated patients are resistant to octreotide. Some patients may present indolent PitNETs detected by screening tests in mutation carriers, who might have a good response to standard treatments [80][55].2.1.4. GPR101
The second known cause of FIPA is due to the germline or somatic microduplication in chromosome Xq26.3, which includes the orphan G-protein-coupled receptor (GPCR) gene, GPR101, a copy number variation (CNV) that is responsible for the so-called X-linked acrogigantism (X-LAG) syndrome first described in 2014 by Trivellin et al. [88][56]. However, there are also sporadic cases that were detected. The c.924G > C (p.E308D) GPR101 missense variant was identified in 4.4% of a series of patients with sporadic acromegaly [89][57]. In the cases that were reported so far, the duplications were germline in females whereas they were somatic in sporadic males with variable levels of mosaicism [90,91][58][59]. However, both sexes had a similar phenotype. These patients were characterized by early childhood (<5 years old in most cases) onset gigantism due to GH-secreting tumors, mixed GH- and PRL-secreting (85% of cases) PitNETs, or hyperplasia [92][60]. There is evidence that pituitary hyperplasia precedes tumor formation in XLAG patients [88][56]. GPR101 encodes a class A, rhodopsin-like orphan GPCR coupled to Gs subunit. Until now, no ligand has been identified as being responsible for the pituitary tumor formation [94][61]. This receptor is normally expressed at the hypothalamus, the nucleus accumbens, and the pituitary gland during fetal life and adolescence. However, relative, scarce, or absent expression is detected during childhood and adult life [89,95][57][62]. The duplication of GPR101 probably affects the GH secretion both at the pituitary and the hypothalamic level. In pituitary tumors harboring a GPR101 duplication, even in the absence of a ligand, the overexpressed GPR101 receptor interacts with the cAMP pathway leading to its constitutive activation and triggering a sequela of proliferative events [88,96][56][63]. However, in one study, it was shown that GPR101 did not constitutively activate the cAMP pathway, while in the same study, GPR101 was also found to inhibit the forskolin-stimulated CRE reporter activity, supporting the fact that it might bind to both stimulatory (Gs) and inhibitory (Gi) proteins [97][64].2.2. MAPK/ERK and PI3K/Akt Pathways
The mitogen-activated protein kinase (MAPK) signaling pathway regulates a variety of physiological processes, such as cell growth, differentiation, and apoptosis, and has been linked to many types of tumors, including lung, prostate, and colorectal cancers [101,102][65][66]. In the MAPK pathway, GTPase Ras is activated by several extracellular growth factors and mitogens after binding to the receptor tyrosine kinases (RTKs) (e.g., IGF-1, EGF, VEGF and FGF receptor families) and the G-protein-coupled receptors (GPCRs). The activated Ras stimulates the protein kinase Raf to phosphorylate and activate MEK and ERK1/2 kinases, which phosphorylate numerous cytoplasmic and nuclear targets, including kinases, phosphatases, and transcription factors (Figure 2). The sustained Ras/ERK signaling has been linked to the upregulation of the genes required for the cell cycle, such as cyclin D1, and the repression of the expression of the genes that inhibit the proliferation, leading to uncontrolled cell proliferation and tumorigenesis [101,102][65][66].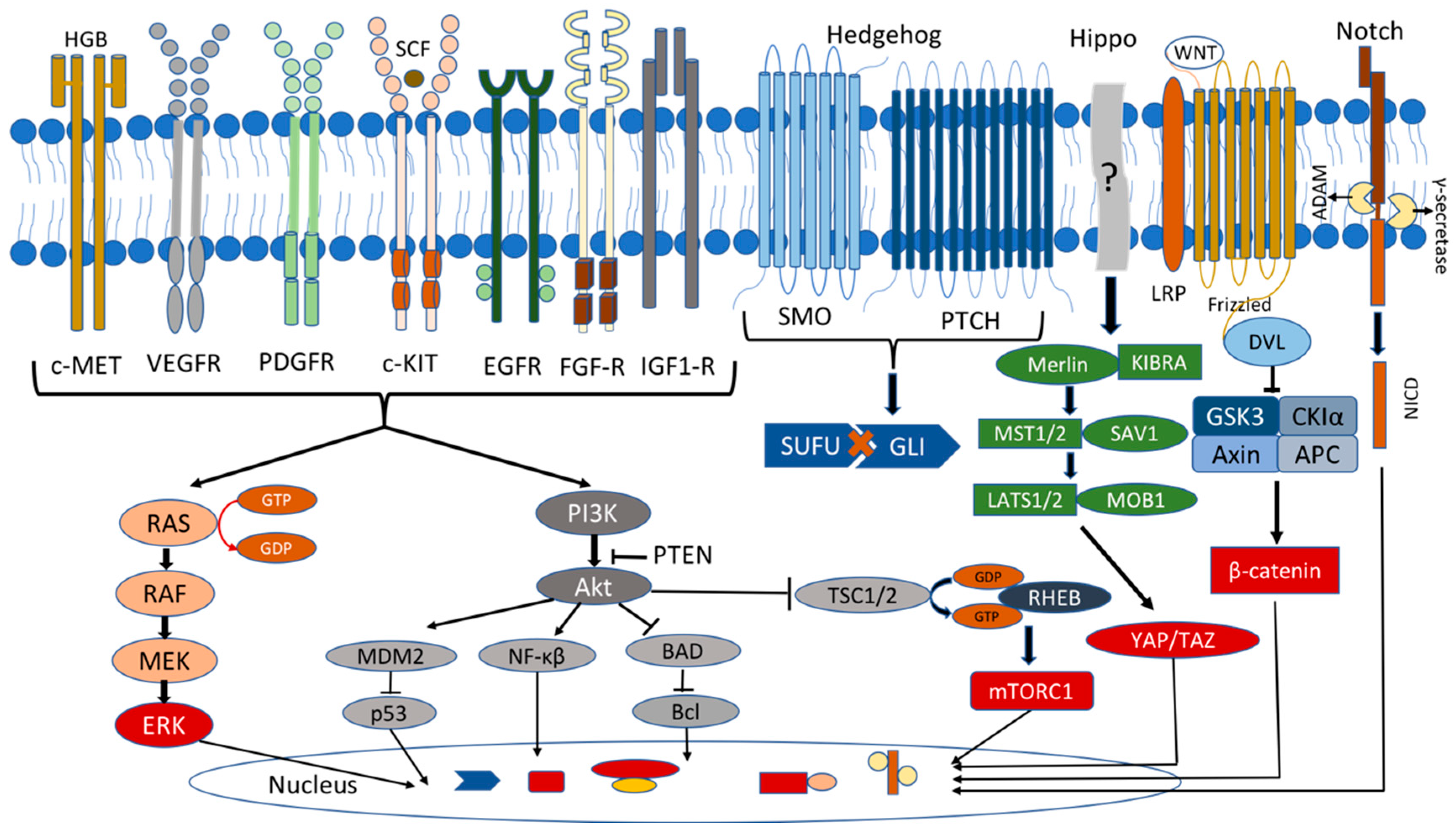
2.3. Hippo Pathway
2.4. Wnt Pathway
Wingless/Int (Wnt) signaling is involved in pituitary organogenesis and controls the cell activity in the adult gland. The Wnt pathway has a pivotal role both in the differentiation of the pluripotent cells and in the proliferation of the mature pituitary cells, as well as in pituitary tumorigenesis. The most crucial component in the intracellular Wnt signaling pathway is β-catenin, an oncogenic protein encoded by the CTNNB1 gene. The Wnt proteins are the crucial regulators of this pathway, which interact with Frizzled (Fzd) receptor and facilitate the transcription of the cell proliferation and differentiation genes. In the inactive state (absence of the Wnt ligand), β-catenin is phosphorylated by the protein complex consisting of AXIN, glycogen synthase kinase-3β (GSK-3β), adenomatosis polyposis coli (APC), and casein kinase 1 alpha (CK1α), leading to its ubiquitination and degradation. In the active state (presence of the Wnt ligand), the regulatory complex axin/APC/GSK-3β/CK1α is inactivated by the disheveled (Dsh) protein, so β-catenin is not phosphorylated and enters the nucleus, acting as a transcription factor for the cell proliferation genes (cyclin D and c-Myc) [213][113]. There is increasing evidence that Wnt signaling is implicated in the PitNets. It has been shown that Wnt4 was highly expressed in human pituitary tumors expressing GH, PRL, and TSH, all of which belong to the Pit1 cell lineage. Its presence was correlated with the Fzd6 expression, suggesting that the activation of the Wnt4/Fzd6 signaling contributed to tumorigenesis, but there was no change in the β-catenin distribution. β-catenin was localized only at the cell membrane in all the pituitary tumors and the normal pituitary glands. These findings indicated that the Wnt4/Fzd6 signaling was activated via a β-catenin-independent pathway [214][114]. Another study investigated 47 pituitary tumors in which β-catenin was localized in the cell membrane with no difference between the PitNETs and normal controls. Still, they found a high nuclear accumulation of the Wnt target genes Cyclin D1 and c-Myc in the tumor tissue, indicating a β-catenin-independent activation of the Wnt pathway [215][115]. Contrary to the previous studies, Semba et al. found a nuclear accumulation of β-catenin in 57% of the investigated PitNets, but they did not compare their findings to the normal pituitary gland [216][116].3. Tumor Suppressor Genes/Oncogenes
3.1. Menin Gene
Multiple endocrine neoplasia type 1 (MEN1) syndrome is an autosomal dominant disorder with a high penetrance that is present in endocrine and non-endocrine tumors. Only 10% of patients are identified with de novo mutations. The patients are predisposed to the formation of the PitNETs, parathyroid hyperplasia, and gastroenteropancreatic neuroendocrine tumors (GEP-NETs) [238][117]. Parathyroid tumors are the most common in approx. 95% of patients, followed by GEP-NETs in approx. 40%. These include gastrinomas, insulinomas, pancreatic polypeptidomas (PPomas), glucagonomas, and vasoactive intestinal polypeptidomas (VIPomas). Anterior pituitary tumors occur in about 30–40% of patients and the most prevalent type is lactortroph tumors (28–80%), followed by NF-PitNETs (15–48.1%), somatotroph tumors (5–15%), co-secreting tumors (9.1%), and rarely corticotroph tumors (5%), depending on the different series [238,239,240][117][118][119]. Overall, MEN1 is responsible for less than 3% of patients with anterior pituitary tumors [241][120]. The causative defect is the germline heterozygous mutation in the MEN1 gene, a tumor suppressor gene localized on chromosome 11q13 [242][121]. Until recently, more than 1200 germline mutations have been identified in the MEN1 gene. In the majority of patients, the tumor formation follows the Knudson’s “two hit model” having one germline mutation in the MEN1 gene while a loss of heterozygosity (LOH) or somatic mutations occurs in the MEN1 alleles of the tumor [243][122]. Menin is a nuclear protein with a ubiquitous expression, which is expressed differently from tissue to tissue [244][123]. The cytoplasmic expression, as well as in the cell membrane, has also been described but to a lesser extent. Menin can regulate the gene transcription either positively or negatively. Recent studies suggest that it may act as a scaffold protein that controls the gene expression and cell signaling [244][123]. Menin binds with the transcription factor JunD, one of the AP-1 transcription factors, and blocks its phosphorylation and activation from the c-Jun N-terminal kinase (JNK). Menin and JunD suppress the expression of the gastrin gene by binding to its promoter [244][123]. On the other hand, menin activates the gene transcription by forming complexes with the transcription activator mixed lineage leukemia protein 1 (MLL1), a methyltransferase which functions as an oncogenic co-factor to promote the gene transcription and leukemogenesis [244,245][123][124].3.2. CDKN1B Gene
Not all patients with a MEN1-like phenotype harbor mutations in menin. About 10–15% have mutations in different genes and 3% of them carry germline mutations in the CDKN1B gene, classified as MEN4 [248][125]. The CDKN1B gene is a tumor suppression gene located on chromosome 12p13.1, encoding for the protein p27Kip1 (known as p27 or as KIP1) [252][126]. The protein p27 is a member of the CDKI family, which binds to the cyclin/cyclin-dependent kinase complexes, preventing the cell cycle progression. In most cases there are germline heterozygous nonsense mutations, which lead to a reduced expression of p27, thereby resulting in an uncontrolled cell cycle proliferation [253][127]. MEN4 patients usually exhibit parathyroid tumors and primary hyperparathyroidism. However, neuroendocrine tumors such as PitNETs, adrenal, and enteropancreatic tumors, testicular and papillary thyroid cancer, as well as non-endocrine tumors such as cervical carcinoma, colon cancer, and meningiomas, have also been reported [253,254][127][128].3.3. CABLES1 (CDK5 and ABL Enzyme Substrate 1)
The CABLES1 gene mapped in the chromosome locus 18q11.2 counteracts the cell cycle progression that is activated in the corticotroph cells in response to glucocorticoids in the adrenal–pituitary negative feedback. The loss-of-function mutations of this tumor suppressor gene leads to an uncontrolled cell proliferation in corticotropinomas [255][129]. The original description of the CABLES1 protein viewed it as an interacting partner and a substrate of the cyclin-dependent kinase-3 (CDK3) [256][130]. In addition, it stabilizes the regulators of the cell cycle, such as CDKN1A (P21), CDK5R1 (P35), and TP63, preventing their degradation [257,258][131][132].3.4. PitNETs Related to Succinate Dehydrogenase (SDHx) Mutations
The SDHx gene mutations are known for their implication in pheochromocytomas and paragagliomas tumor formation [259][133]. However, in 2012, Xekouki et al. described a patient with an acromegaly and concomitant presence of paragangliomas (PGLs) and pheochromocytomas (PHEOs) carrying a germline SDHD mutation while he exhibited loss of heterozygosity at the SDHD locus in the pituitary tumor, and increased transcription hypoxia-inducible factor α(HIF-1α) levels similar to the PHEO/PGLs [260][134]. Subsequently, the same group described the 3PAs syndrome characterized by the presence of the PHEOs and/or PGLs, and pituitary adenoma in the same patient [261][135]. Although the SDHx mutations are common in the 3PAs familiar cases (62.5–75%), they are quite rare in the sporadic setting of the syndrome (0.3–1.8%) [261,262][135][136]. The SDHx genes are tumor suppressor genes, encoding for the different subunits of the mitochondrial enzyme SDH, also named complex II or succinate:quinone oxidoreductase [263][137]. SDH is located in the inner mitochondrial membrane and has a critical role in the oxidative phosphorylation (OXPHOS) and tricarboxylic acid (TCA) cycles, two major mechanisms in the metabolism and energy production within the cells [264][138]. SDH consists of four subunits, SDHA-D. SDHA and B constitute the catalytic domain, which is extrinsic on the matrix side, while SDHC and D comprise the anchor subunits, which are intrinsic transmembrane proteins. The catalytic subunits catalyze the oxidation of the succinate to fumarate while the anchor subunits contribute to the transfer of the electrons from the succinate in the mitochondrial matrix to the ubiquinone in the inner membrane [264][138]. The PitNETs in the 3PAs are more common among familial cases and they are usually macroadenomas secreting PRL or GH, while less frequently, they can be non-functioning and secrete ACTH [269][139]. Most of the described cases required more than one type of treatment as they exhibited a more aggressive behavior and resistance to SSAs. Interestingly, the PitNETs in the context of the 3PAs were present at a younger age, in contrast to non-syndromic pituitary tumors, while the co-existence with the PHEO/PGLs was compatible with a more aggressive pituitary tumor, which implies a critical role of these tumors in the phenotype of the disease [267,269][139][140].3.5. DICER1, Ribonuclease III
DICER1 is a predisposition syndrome for the different types of tumors characterized by germline or mosaic loss-of-function (LOF) mutations in the DICER1 gene mapped on the chromosome locus 14q32.13 [274][141]. It encodes a ubiquitously expressed endonuclease, a member of the ribonuclease (RNase) III family, required for the biogenesis of microRNA (miRNA) and small interfering RNA V (siRNA). However, the specific role of the DICER1 gene in pituitary tumorigenesis is still under investigation [274,275][141][142]. The most characteristic tumor in DICER1 patients is pleuropulmonary blastoma (PBB), a rare, early childhood pulmonary mesenchyma tumor. The other tumors include cystic nephroma, Wilms tumors, ovarian sex cord-stromal tumors (OSCSTs), especially Sertoli–Leydig cell tumors (SLCTs), and childhood embryonal rhabdomyosarcomas (ERMS) [276][143]. Pituitary blastoma, a very rare embryonal aggressive pituitary tumor, can be part of DICER1 expressed with an ACTH-dependent hypercortisolemia (Cushing disease) and neuro-ophthalmopathy. Apart from surgery, polychemotherapy (cyclophosphamide, vincristine, methotrexate, carboplatin, and etoposide used in DICER1 patients) and adjuvant radiotherapy may be needed. However, the clinical experience with such tumors is very limited [277][144].4. Stem Cells in the Pituitary Gland and Tumorigenesis
Nowadays, it is well established that cancer stem cells (CSCs) stimulate tumor initiation, progression, recurrence, metastasis, and/or therapy resistance in different types of tumors. CSCs are characterized by persistent self-renewal and a multipotent differentiation capacity, representing a tumor-initiating cell population with intra-tumor heterogeneity [294][145]. Additionally, CSCs have high levels of plasticity with the ability to dedifferentiate. Similarly, CSCs have been identified in PitNETs. Several studies have isolated CSCs from human pituitary tumors with a clonogenic, sphere-forming potential in cultures that expressed pituitary-specific markers, such as Pit1, and markers of stemness, such as OCT4, Notch1 and 4, CD15, CD90, CD133, NESTIN, NANOG, CXCR4, and KLF4 [295,296,297,298,299,300][146][147][148][149][150][151]. Additionally, the regulatory signaling pathways that are essential for self-renewal and the differentiation of normal stem cells, such as Notch, Sonic hedgehog, Wnt, and Hippo are associated with cancer stem cells and pituitary oncogenesis as well [109][73].
Moreover, recent studies suggested that human pituitary adenoma stem cells (hPASCs) express DRD2, SSTR2, and SSTR5, whose activation using current treatment strategies such as DAs and SSAs seem to have promising results [296,297][147][148]. For example, Würth and his colleagues showed a decreased cell survival in hPASC cultures when incubated using the somatostatin/dopamine chimera BIM-23A760 [296][147]. Similarly, another study demonstrated that the DRD2 agonist BIM53097 and SSTR2 agonist BIM23120 had antiproliferative effects on both the spheres and tumor tissues in about half of the studied NF-PitNETs. In addition, the reduction in the proliferation ability of sphere-forming cells was confirmed by an increased CDKI p27 expression and a decrease in the cyclin D3 expression [297][148]. It is important to note that there was no difference in the frequency of the sphere formation between the NF-PitNETs that were in vitro resistant or sensitive to DRD2 and the SSTR2 agonists. However, the spheres that came from the tumors resistant to the DRD2 and SSTR2 agonists were larger compared to those derived from the sensitive NF-PitNETs [297][148].
5. MicroRNAs
MicroRNAs are short protein non-coding RNAs that act as regulatory proteins and control the post-transcriptional expression of specific genes through RNA interference and mRNA destabilization. They can induce a rapid degradation of the target messenger or inhibit its translation into a protein, and their expression can be regulated at different levels [302][152]. In 2005, their expression was described for the first time in the pituitary gland. Since then, several studies have shown that miRNAs are involved in many mechanisms regulating the pituitary hormone production, tumor formation, progression, and aggressiveness [303,304,305][153][154][155]. MiRNAs may play an important role in the pathogenesis and progression of PitNETs and may provide new molecular targets for their diagnosis and treatment. It is estimated that miRNAs may control up to 50% of all the protein-coding genes [306][156]. Several miRNAs are found to be involved in cell proliferation and apoptosis through an interference with the different pathways. For instance, the miR-187-3p elevation seems to promote the cell cycle progression and inhibit the proliferation of pituitary tumor cells via the NF-κB signaling pathway [307][157]. Furthermore, the upregulation of several miRNAs (miR-17-5p, miR-20a, miR-106b, miR-21, miR200c, and miR-128) in pituitary tumors may inhibit the tumor suppressor signaling pathway PIK3/AKT, including PTEN, enabling a more aggressive behavior of these tumors [302,308][152][158]. On the other hand, another group of miRNAs (miR-132, miR-15a, and miR-16) has the ability to inhibit the cell invasion and metastasis in several PitNETs by targeting SOX5, rendering these miRNAs as potential therapeutic targets for more aggressive pituitary tumors [309][159].References
- Banskota, S.; Adamson, D.C. Pituitary Adenomas: From Diagnosis to Therapeutics. Biomedicines 2021, 9, 494.
- Daly, A.F.; Beckers, A. The Epidemiology of Pituitary Adenomas. Endocrinol. Metab. Clin. 2020, 49, 347–355.
- Teramoto, A.; Hirakawa, K.; Sanno, N.; Osamura, Y. Incidental pituitary lesions in 1,000 unselected autopsy specimens. Radiology 1994, 193, 161–164.
- Vallecillos, F.J.T.; Fernández, S.O. Histopathological features of post-mortem pituitaries: A retrospective analysis. Rev. Assoc. Med. Bras. 2016, 62, 399–406.
- Molitch, M.E. Diagnosis and treatment of pituitary adenomas: A review. JAMA J. Am. Med. Assoc. 2017, 317, 516–524.
- Biermasz, N.R. The burden of disease for pituitary patients. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101309.
- Asa, S.L.; Mete, O.; Perry, A.; Osamura, R.Y. Overview of the 2022 WHO Classification of Pituitary Tumors. Endocr. Pathol. 2022, 33, 6–26.
- Ershadinia, N.; Dsc, N.A.T. Diagnosis and Treatment of Acromegaly: An Update. Mayo Clin. Proc. 2022, 97, 333–346.
- Lasolle, H.; Ferriere, A.; Vasiljevic, A.; Eimer, S.; Nunes, M.L.; Tabarin, A. Pasireotide-LAR in acromegaly patients treated with a combination therapy: A real-life study. Endocr. Connect. 2019, 8, 1383–1394.
- Inder, W.J.; Jang, C. Treatment of Prolactinoma. Medicina 2022, 58, 1095.
- Yavropoulou, M.P.; Yavropoulou, M.P. The natural history and treatment of non-functioning pituitary adenomas (non-functioning PitNETs). Endocr.-Relat. Cancer 2020, 27, R375–R390.
- Daly, A.F.; Bch, M.B.; Tichomirowa, M.A.; Beckers, A. Best Practice & Research Clinical Endocrinology & Metabolism The epidemiology and genetics of pituitary adenomas. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 543–554.
- Dénes, J.; Korbonits, M. The clinical aspects of pituitary tumour genetics. Endocrine 2021, 71, 663–674.
- Peculis, R.; Niedra, H.; Rovite, V. Large scale molecular studies of pituitary neuroendocrine tumors: Novel markers, mechanisms and translational perspectives. Cancers 2021, 13, 1395.
- Xekouki, P.; Azevedo, M.; Stratakis, C.A. Anterior pituitary adenomas: Inherited syndromes, novel genes and molecular pathways. Expert Rev. Endocrinol. Metab. 2010, 5, 697–709.
- Xekouki, P.; Lodge, E.J.; Matschke, J.; Santambrogio, A.; Apps, J.R.; Sharif, A.; Jacques, T.S.; Aylwin, S.; Prevot, V.; Li, R.; et al. Non-secreting pituitary tumours characterised by enhanced expression of YAP/TAZ. Endocr. Relat. Cancer 2019, 26, 215–225.
- Russell, J.P.; Lodge, E.J.; Andoniadou, C.L. Basic Research Advances on Pituitary Stem Cell Function and Regulation. Neuroendocrinology 2018, 107, 196–203.
- Donati, S.; Aurilia, C.; Palmini, G.; Miglietta, F.; Falsetti, I.; Iantomasi, T.; Brandi, M.L. Micrornas as potential biomarkers in pituitary adenomas. Non-Coding RNA 2021, 7, 55.
- SUTHERLAND, E.W.; RALL, T.W. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J. Biol. Chem. 1958, 232, 1077–1091.
- Milligan, G.; Kostenis, E. Heterotrimeric G-proteins: A short history. Br. J. Pharmacol. 2006, 147, S46–S55.
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650.
- Hernández-Ramírez, L.C.; Trivellin, G.; Stratakis, C.A. Cyclic 3′,5′-adenosine monophosphate (cAMP) signaling in the anterior pituitary gland in health and disease. Mol. Cell. Endocrinol. 2018, 463, 72–86.
- Vallar, L.; Spada, A.; Giannattasio, G. Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas. Nature 1987, 330, 566–568.
- Mantovani, G.; Ballare, E.; Giammona, E.; Beck-Peccoz, P.; Spada, A. The Gsα gene: Predominant maternal origin of transcription in human thyroid gland and gonads. J. Clin. Endocrinol. Metab. 2002, 87, 4736–4740.
- Bastepe, M.; Jüppner, H. GNAS locus and pseudohypoparathyroidism. Horm. Res. 2005, 63, 65–74.
- Campbell, R.; Gosden, C.M.; Bonthron, D.T. Parental origin of transcription from the human GNAS1 gene. J. Med. Genet. 1994, 31, 607–614.
- Landis, C.A.; Masters, S.B.; Spada, A.; Pace, A.M.; Bourne, H.R.; Vallar, L. GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 1989, 340, 692–696.
- Riminucci, M.; Collins, M.T.; Lala, R.; Corsi, A.; Matarazzo, P.; Robey, P.G.; Bianco, P. An R201H activating mutation of the GNAS1 (Gsα) gene in a corticotroph pituitary adenoma. J. Clin. Pathol.-Mol. Pathol. 2002, 55, 58–60.
- Taboada, G.F.; Tabet, A.L.O.; Naves, L.A.; Carvalho, D.P.; Gadelha, M.R. Prevalence of gsp oncogene in somatotropinomas and clinically non-functioning pituitary adenomas: Our experience. Pituitary 2009, 12, 165–169.
- Pertuit, M.; Barlier, A.; Enjalbert, A.; Gérard, C. Signalling pathway alterations in pituitary adenomas: Involvement of Gsα, cAMP and mitogen-activated protein kinases. J. Neuroendocrinol. 2009, 21, 869–877.
- Theodoropoulou, M.; Stalla, G.K. Somatostatin receptors: From signaling to clinical practice. Front. Neuroendocrinol. 2013, 34, 228–252.
- Albright, F.; Butler, A.M.; Hampton, A.O.; Smith, P. Disseminata, Areas of Pigmentation and Endocrine. N. Engl. J. Med. 1937, 216, 721–746.
- Dumitrescu, C.E.; Collins, M.T. McCune-Albright syndrome. Orphanet J. Rare Dis. 2008, 3, 12.
- Weinstein, L.S.; Shenker, A.; Gejman, P.V.; Merino, M.J.; Friedman, E.; Spiegel, A.M. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N. Engl. J. Med. 1991, 325, 1688–1695.
- Salenave, S.; Boyce, A.M.; Collins, M.T.; Chanson, P. Acromegaly and mccune-albright syndrome. J. Clin. Endocrinol. Metab. 2014, 99, 1955–1969.
- Akintoye, S.O.; Kelly, M.H.; Brillante, B.; Cherman, N.; Turner, S.; Butman, J.A.; Robey, P.G.; Collins, M.T. Pegvisomant for the treatment of gsp-mediated growth hormone excess in patients with McCune-Albright syndrome. J. Clin. Endocrinol. Metab. 2006, 91, 2960–2966.
- Correa, R.; Salpea, P.; Stratakis, C.A. Carney complex: An update. Eur. J. Endocrinol. 2015, 173, M85–M97.
- Bouys, L.; Bertherat, J. Carney complex: Clinical and genetic update 20 years after the identification of the CNC1 (PRKAR1A) gene. Eur. J. Endocrinol. 2021, 184, R99–R109.
- Courcoutsakis, N.A.; Tatsi, C.; Patronas, N.J.; Lee, C.C.R.; Prassopoulos, P.K.; Stratakis, C.A. The complex of myxomas, spotty skin pigmentation and endocrine overactivity (Carney complex): Imaging findings with clinical and pathological correlation. Insights Imaging 2013, 4, 119–133.
- Stergiopoulos, S.G.; Abu-Asab, M.S.; Tsokos, M.; Stratakis, C.A. Pituitary pathology in Carney complex patients. Pituitary 2004, 7, 73–82.
- Kirschner, L.S. PRKAR1A and the evolution of pituitary tumors. Mol. Cell. Endocrinol. 2010, 326, 3–7.
- Kiefer, F.W.; Winhofer, Y.; Iacovazzo, D.; Korbonits, M.; Wolfsberger, S.; Knosp, E.; Trautinger, F.; Höftberger, R.; Krebs, M.; Luger, A.; et al. PRKAR1A mutation causing pituitary-dependent Cushing disease in a patient with Carney complex. Eur. J. Endocrinol. 2017, 177, K7–K12.
- Hernández-Ramírez, L.C.; Tatsi, C.; Lodish, M.B.; Faucz, F.R.; Pankratz, N.; Chittiboina, P.; Lane, J.; Kay, D.M.; Valdés, N.; Dimopoulos, A.; et al. Corticotropinoma as a component of carney complex. J. Endocr. Soc. 2017, 1, 918–925.
- Hernández-Ramírez, L.C.; Gabrovska, P.; Dénes, J.; Stals, K.; Trivellin, G.; Tilley, D.; Ferraù, F.; Evanson, J.; Ellard, S.; Grossman, A.B.; et al. Landscape of familial isolated and young-onset pituitary adenomas: Prospective diagnosis in AIP mutation carriers. J. Clin. Endocrinol. Metab. 2015, 100, E1242–E1254.
- Tiryakioǧlu, Ö.; Caneroǧlu, N.Ü.; Yilmaz, E.; Gazioǧlu, N.; Kadioǧlu, P.; Açbay, Ö.; Gündoǧdu, S. Familial acromegaly: A familial report and review of the literature. Endocr. Res. 2004, 30, 239–245.
- Vierimaa, O.; Georgitsi, M.; Lehtonen, R.; Vahteristo, P.; Kokko, A.; Raitila, A.; Tuppurainen, K.; Ebeling, T.M.L.; Salmela, P.I.; Paschke, R.; et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science 2006, 312, 1228–1230.
- Daly, A.F.; Tichomirowa, M.A.; Petrossians, P.; Heliövaara, E.; Jaffrain-Rea, M.L.; Barlier, A.; Naves, L.A.; Ebeling, T.; Karhu, A.; Raappana, A.; et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: An international collaborative study. J. Clin. Endocrinol. Metab. 2010, 95, 373–383.
- Daly, A.F.; Beckers, A. The role of AIP mutations in pituitary adenomas: 10 years on. Endocrine 2016, 55, 333–335.
- Trivellin, G.; Korbonits, M. AIP and its interacting partners. J. Endocrinol. 2011, 210, 137–155.
- Formosa, R.; Xuereb-Anastasi, A.; Vassallo, J. Aip regulates cAMP signalling and GH secretion in GH3 cells. Endocr. Relat. Cancer 2013, 20, 495–505.
- Hernández-Ramírez, L.C.; Trivellin, G.; Stratakis, C.A. Role of Phosphodiesterases on the Function of Aryl Hydrocarbon Receptor-Interacting Protein (AIP) in the Pituitary Gland and on the Evaluation of AIP Gene Variants. Horm. Metab. Res. 2017, 49, 286–295.
- De Pinho, L.K.J.; Neto, L.V.; Wildemberg, L.E.A.; Gasparetto, E.L.; Marcondes, J.; De Almeida Nunes, B.; Takiya, C.M.; Gadelha, M.R. Low aryl hydrocarbon receptor-interacting protein expression is a better marker of invasiveness in somatotropinomas than Ki-67 and p53. Neuroendocrinology 2011, 94, 39–48.
- Chahal, H.S.; Trivellin, G.; Leontiou, C.A.; Alband, N.; Fowkes, R.C.; Tahir, A.; Igreja, S.C.; Chapple, J.P.; Jordan, S.; Lupp, A.; et al. Somatostatin analogs modulate AIP in somatotroph adenomas: The role of the ZAC1 pathway. J. Clin. Endocrinol. Metab. 2012, 97, E1411–E1420.
- Dutta, P.; Reddy, K.S.; Rai, A.; Madugundu, A.K.; Solanki, H.S.; Bhansali, A.; Radotra, B.D.; Kumar, N.; Collier, D.; Iacovazzo, D.; et al. Surgery, Octreotide, Temozolomide, Bevacizumab, Radiotherapy, and Pegvisomant Treatment of an AIP Mutation—Positive Child. J. Clin. Endocrinol. Metab. 2019, 104, 3539–3544.
- Williams, F.; Hunter, S.; Bradley, L.; Chahal, H.S.; Storr, H.L.; Akker, S.A.; Kumar, A.V.; Orme, S.M.; Evanson, J.; Abid, N.; et al. Clinical experience in the screening and management of a large kindred with familial isolated pituitary adenoma due to an aryl hydrocarbon receptor interacting protein (AIP) mutation. J. Clin. Endocrinol. Metab. 2014, 99, 1122–1131.
- Trivellin, G.; Daly, A.F.; Faucz, F.R.; Yuan, B.; Rostomyan, L.; Larco, D.O.; Schernthaner-Reiter, M.H.; Szarek, E.; Leal, L.F.; Caberg, J.-H.; et al. Gigantism and Acromegaly Due to Xq26 Microduplications and GPR101 Mutation. N. Engl. J. Med. 2014, 371, 2363–2374.
- Trivellin, G.; Hernández-Ramírez, L.C.; Swan, J.; Stratakis, C.A. An orphan G-protein-coupled receptor causes human gigantism and/or acromegaly: Molecular biology and clinical correlations. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 125–140.
- Daly, A.F.; Yuan, B.; Fina, F.; Caberg, J.H.; Trivellin, G.; Rostomyan, L.; De Herder, W.W.; Naves, L.A.; Metzger, D.; Cuny, T.; et al. Somatic mosaicism underlies X-linked acrogigantism syndrome in sporadic male subjects. Endocr. Relat. Cancer 2016, 23, 221–233.
- Iacovazzo, D.; Caswell, R.; Bunce, B.; Jose, S.; Yuan, B.; Hernández-Ramírez, L.C.; Kapur, S.; Caimari, F.; Evanson, J.; Ferraù, F.; et al. Germline or somatic GPR101 duplication leads to X-linked acrogigantism: A clinico-pathological and genetic study. Acta Neuropathol. Commun. 2016, 4, 56.
- Daly, A.F.; Lysy, P.A.; Desfilles, C.; Rostomyan, L.; Mohamed, A.; Caberg, J.H.; Raverot, V.; Castermans, E.; Marbaix, E.; Maiter, D.; et al. GHRH excess and blockade in X-LAG syndrome. Endocr. Relat. Cancer 2016, 23, 161–170.
- Alexander, S.P.H.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G protein-coupled receptors. Br. J. Pharmacol. 2019, 176, S21–S141.
- Trivellin, G.; Bjelobaba, I.; Daly, A.F.; Larco, D.O.; Palmeira, L.; Faucz, F.R.; Thiry, A.; Leal, L.F.; Rostomyan, L.; Quezado, M.; et al. Characterization of GPR101 transcript structure and expression patterns. J. Mol. Endocrinol. 2016, 57, 97–111.
- Trivellin, G.; Faucz, F.R.; Daly, A.F.; Beckers, A.; Stratakis, C.A. Hereditary endocrine tumours: Current state-of-the-art and research opportunities: GPR101, an orphan GPCR with roles in growth and pituitary tumorigenesis. Endocr. Relat. Cancer 2020, 27, T87–T97.
- Martin, A.L.; Steurer, M.A.; Aronstam, R.S. Constitutive Activity among Orphan Class-A G Protein Coupled Receptors. PLoS ONE 2015, 10, e0138463.
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290.
- Cakir, M.; Grossman, A.B. Targeting MAPK (Ras/ERK) and PI3K/Akt pathways in pituitary tumorigenesis. Expert Opin. Ther. Targets 2009, 13, 1121–1134.
- Hou, X.Z.; Liu, W.; Fan, H.T.; Liu, B.; Pang, B.; Xin, T.; Xu, S.C.; Pang, Q. Expression of hepatocyte growth factor and its receptor c-Met in human pituitary adenomas. Neuro. Oncol. 2010, 12, 799.
- Casar-Borota, O.; Fougner, S.L.; Bollerslev, J.; Nesland, J.M. KIT protein expression and mutational status of KIT gene in pituitary adenomas. Virchows Arch. 2012, 460, 171–181.
- Kowarik, M.; Onofri, C.; Colaco, T.; Stalla, G.K.; Renner, U. Platelet-derived growth factor (PDGF) and PDGF receptor expression and function in folliculostellate pituitary cells. Exp. Clin. Endocrinol. Diabetes 2010, 118, 113–120.
- Monsalves, E.; Juraschka, K.; Tateno, T.; Agnihotri, S.; Asa, S.L.; Ezzat, S.; Zadeh, G. The PI3K/AKT/mTOR pathway in the pathophysiology and treatment of pituitary adenomas. Endocr. Relat. Cancer 2014, 21, R331–R344.
- Lodge, E.J.; Santambrogio, A.; Russell, J.P.; Xekouki, P.; Jacques, T.S.; Johnson, R.L.; Thavaraj, S.; Bornstein, S.R.; Andoniadou, C.L. Homeostatic and tumourigenic activity of SOX2 + pituitary stem cells is controlled by the LATS/YAP/TAZ cascade. Elife 2019, 8, e43996.
- Gaston-Massuet, C.; Andoniadou, C.L.; Signore, M.; Jayakody, S.A.; Charolidi, N.; Kyeyune, R.; Vernay, B.; Jacques, T.S.; Taketo, M.M.; Le Tissier, P.; et al. Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc. Natl. Acad. Sci. USA 2011, 108, 11482–11487.
- Cox, B.; Roose, H.; Vennekens, A.; Vankelecom, H. Pituitary stem cell regulation: Who is pulling the strings? J. Endocrinol. 2017, 234, R135–R158.
- Gomes, D.C.; Jamra, S.A.; Leal, L.F.; Colli, L.M.; Campanini, M.L.; Oliveira, R.S.; Martinelli, C.E.; Moreira, A.C.; Machado, H.R.; Saggioro, F.; et al. Sonic Hedgehog pathway is upregulated in adamantinomatous craniopharyngiomas. Eur. J. Endocrinol. 2015, 172, 603–608.
- Karga, H.J.; Alexander, J.M.; Hedley-Whyte, E.T.; Klibanski, A.; Jameson, J.L. Ras mutations in human pituitary tumors. J. Clin. Endocrinol. Metab. 1992, 74, 914–919.
- Cai, W.Y.; Alexander, J.M.; Hedley-Whyte, E.T.; Scheithauer, B.W.; Jameson, J.L.; Zervas, N.T.; Klibanski, A. Ras mutations in human prolactinomas and pituitary carcinomas. J. Clin. Endocrinol. Metab. 1994, 78, 89–93.
- Ewing, I.; Pedder-Smith, S.; Franchi, G.; Ruscica, M.; Emery, M.; Vax, V.; Garcia, E.; Czirják, S.; Hanzély, Z.; Kola, B.; et al. A mutation and expression analysis of the oncogene BRAF in pituitary adenomas. Clin. Endocrinol. 2007, 66, 348–352.
- Dworakowska, D.; Wlodek, E.; Leontiou, C.A.; Igreja, S.; Cakir, M.; Teng, M.; Prodromou, N.; Góth, M.I.; Grozinsky-Glasberg, S.; Gueorguiev, M.; et al. Activation of RAF/MEK/ERK and PI3K/AKT/mTOR pathways in pituitary adenomas and their effects on downstream effectors. Endocr. Relat. Cancer 2009, 16, 1329–1338.
- Chaturvedi, K.; Sarkar, D.K. Mediation of basic fibroblast growth factor-induced lactotropic cell proliferation by Src-Ras-mitogen-activated protein kinase p44/42 signaling. Endocrinology 2005, 146, 1948–1955.
- Oomizu, S.; Chaturvedi, K.; Sarkar, D.K. Folliculostellate cells determine the susceptibility of lactotropes to estradiol’s mitogenic action. Endocrinology 2004, 145, 1473–1480.
- Booth, A.; Trudeau, T.; Gomez, C.; Scott Lucia, M.; Gutierrez-Hartmann, A. Persistent ERK/MAPK activation promotes lactotrope differentiation and diminishes tumorigenic phenotype. Mol. Endocrinol. 2014, 28, 1999–2011.
- Aylwin, S.J.B.; Bodi, I.; Beaney, R. Pronounced response of papillary craniopharyngioma to treatment with vemurafenib, a BRAF inhibitor. Pituitary 2016, 19, 544–546.
- Chik, C.L.; Van Landeghem, F.K.H.; Easaw, J.C.; Mehta, V. Aggressive Childhood-onset Papillary Craniopharyngioma Managed With Vemurafenib, a BRAF Inhibitor. J. Endocr. Soc. 2021, 5, bvab043.
- Himes, B.T.; Ruff, M.W.; Van Gompel, J.J.; Park, S.S.; Galanis, E.; Kaufmann, T.J.; Uhm, J.H. Recurrent papillary craniopharyngioma with BRAF V600E mutation treated with dabrafenib: Case report. J. Neurosurg. 2018, 130, 1299–1303.
- Rao, M.; Bhattacharjee, M.; Shepard, S.; Hsu, S. Newly diagnosed papillary craniopharyngioma with BRAF V600E mutation treated with single-agent selective BRAF inhibitor dabrafenib: A case report. Oncotarget 2019, 10, 6038–6042.
- Bernstein, A.; Mrowczynski, O.D.; Greene, A.; Ryan, S.; Chung, C.; Zacharia, B.E.; Glantz, M. Dual BRAF/MEK therapy in BRAF V600E-mutated primary brain tumors: A case series showing dramatic clinical and radiographic responses and a reduction in cutaneous toxicity. J. Neurosurg. 2019, 133, 1704–1709.
- Khaddour, K.; Chicoine, M.R.; Huang, J.; Dahiya, S.; Ansstas, G. Successful Use of BRAF/MEK Inhibitors as a Neoadjuvant Approach in the Definitive Treatment of Papillary Craniopharyngioma. J. Natl. Compr. Cancer Netw. 2020, 18, 1590–1595.
- Di Stefano, A.L.; Guyon, D.; Sejean, K.; Feuvret, L.; Villa, C.; Berzero, G.; Desforges Bullet, V.; Halimi, E.; Boulin, A.; Baussart, B.; et al. Medical debulking with BRAF/MEK inhibitors in aggressive BRAF-mutant craniopharyngioma. Neuro-Oncol. Adv. 2020, 2, vdaa141.
- Calvanese, F.; Jacquesson, T.; Manet, R.; Vasiljevic, A.; Lasolle, H.; Ducray, F.; Raverot, G.; Jouanneau, E. Neoadjuvant B-RAF and MEK Inhibitor Targeted Therapy for Adult Papillary Craniopharyngiomas: A New Treatment Paradigm. Front. Endocrinol. 2022, 13, 882381.
- Brastianos, P.K.; Twohy, E.; Geyer, S.M.; Gerstner, E.R.; Kaufmann, T.J.; Ruff, M.; Bota, D.A.; Reardon, D.A.; Cohen, A.L.; La Fuente, M.I.D.; et al. Alliance A071601: Phase II trial of BRAF/MEK inhibition in newly diagnosed papillary craniopharyngiomas. J. Clin. Oncol. 2021, 39, 2000.
- Stork, P.J.S.; Schmitt, J.M. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002, 12, 258–266.
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328.
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 2012, 16 (Suppl. 2), S17–S27.
- Reubi, J.C.; Laissue, J.A. Multiple actions of somatostatin in neoplastic disease. Trends Pharmacol. Sci. 1995, 16, 110–115.
- Bruns, C.; Lewis, I.; Briner, U.; Meno-Tetang, G.; Weckbecker, G. SOM230: A novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur. J. Endocrinol. 2002, 146, 707–716.
- Weckbecker, G.; Briner, U.; Lewis, I.; Bruns, C. SOM230: A new somatostatin peptidomimetic with potent inhibitory effects on the growth hormone/insulin-like growth factor-I axis in rats, primates, and dogs. Endocrinology 2002, 143, 4123–4130.
- Hubina, E.; Nanzer, A.M.; Hanson, M.R.; Ciccarelli, E.; Losa, M.; Gaia, D.; Papotti, M.; Terreni, M.R.; Khalaf, S.; Jordan, S.; et al. Somatostatin analogues stimulate p27 expression and inhibit the MAP kinase pathway in pituitary tumours. Eur. J. Endocrinol. 2006, 155, 371–379.
- Theodoropoulou, M.; Zhang, J.; Laupheimer, S.; Paez-Pereda, M.; Erneux, C.; Florio, T.; Pagotto, U.; Stalla, G.K. Octreotide, a somatostatin analogue, mediates its antiproliferative action in pituitary tumor cells by altering phosphatidylinositol 3-kinase signaling and inducing Zac1 expression. Cancer Res. 2006, 66, 1576–1582.
- Liu, J.C.; Baker, R.E.; Sun, C.; Sundmark, V.C.; Elsholtz, H.P. Activation of Go-coupled dopamine D2 receptors inhibits ERK1/ERK2 in pituitary cells. A key step in the transcriptional suppression of the prolactin gene. J. Biol. Chem. 2002, 277, 35819–35825.
- Iaccarino, C.; Samad, T.A.; Mathis, C.; Kercret, H.; Picetti, R.; Borrelli, E. Control of lactotrop proliferation by dopamine: Essential role of signaling through D2 receptors and ERKs. Proc. Natl. Acad. Sci. USA 2002, 99, 14530–14535.
- Radl, D.; De Mei, C.; Chen, E.; Lee, H.; Borrelli, E. Each individual isoform of the dopamine D2 receptor protects from lactotroph hyperplasia. Mol. Endocrinol. 2013, 27, 953–965.
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31.
- Ilie, M.D.; Lasolle, H.; Raverot, G. Emerging and Novel Treatments for Pituitary Tumors. J. Clin. Med. 2019, 8, 1107.
- Beuvink, I.; Boulay, A.; Fumagalli, S.; Zilbermann, F.; Ruetz, S.; O’Reilly, T.; Natt, F.; Hall, J.; Lane, H.A.; Thomas, G. The mTOR inhibitor RAD001 sensitizes tumor cells to DNA-damaged induced apoptosis through inhibition of p21 translation. Cell 2005, 120, 747–759.
- Gorshtein, A.; Rubinfeld, H.; Kendler, E.; Theodoropoulou, M.; Cerovac, V.; Stalla, G.K.; Cohen, Z.R.; Hadani, M.; Shimon, I. Mammalian target of rapamycin inhibitors rapamycin and RAD001 (everolimus) induce anti-proliferative effects in GH-secreting pituitary tumor cells in vitro. Endocr. Relat. Cancer 2009, 16, 1017–1027.
- Zatelli, M.C.; Minoia, M.; Filieri, C.; Tagliati, F.; Buratto, M.; Ambrosio, M.R.; Lapparelli, M.; Scanarini, M.; Degli Uberti, E.C. Effect of everolimus on cell viability in nonfunctioning pituitary adenomas. J. Clin. Endocrinol. Metab. 2010, 95, 968–976.
- Cerovac, V.; Monteserin-Garcia, J.; Rubinfeld, H.; Buchfelder, M.; Losa, M.; Florio, T.; Paez-Pereda, M.; Stalla, G.K.; Theodoropoulou, M. The somatostatin analogue octreotide confers sensitivity to rapamycin treatment on pituitary tumor cells. Cancer Res. 2010, 70, 666–674.
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828.
- Lodge, E.J.; Russell, J.P.; Patist, A.L.; Francis-West, P.; Andoniadou, C.L. Expression analysis of the Hippo cascade indicates a role in pituitary stem cell development. Front. Physiol. 2016, 7, 114.
- Zheng, Y.; Pan, D. The Hippo Signaling Pathway in Development and Disease. Dev. Cell 2019, 50, 264–282.
- Basu-Roy, U.; Bayin, N.S.; Rattanakorn, K.; Han, E.; Placantonakis, D.G.; Mansukhani, A.; Basilico, C. Sox2 antagonizes the Hippo pathway to maintain stemness in cancer cells. Nat. Commun. 2015, 6, 6411.
- St John, M.A.R.; Tao, W.; Fei, X.; Fukumoto, R.; Carcangiu, M.L.; Brownstein, D.G.; Parlow, A.F.; McGrath, J.; Xu, T. Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat. Genet. 1999, 21, 182–186.
- Iglesias, P. Targeted therapies in the medical management of craniopharyngioma. Pituitary 2022, 25, 383–392.
- Miyakoshi, T.; Takei, M.; Kajiya, H.; Egashira, N.; Takekoshi, S.; Teramoto, A.; Osamura, R.Y. Expression of Wnt4 in human pituitary adenomas regulates activation of the beta-catenin-independent pathway. Endocr. Pathol. 2008, 19, 261–273.
- Formosa, R.; Gruppetta, M.; Falzon, S.; Santillo, G.; DeGaetano, J.; Xuereb-Anastasi, A.; Vassallo, J. Expression and clinical significance of Wnt players and survivin in pituitary tumours. Endocr. Pathol. 2012, 23, 123–131.
- Semba, S.; Han, S.-Y.; Ikeda, H.; Horii, A. Frequent Nuclear Accumulation of-Catenin in Pituitary Adenoma. Cancer 2001, 91, 42–48.
- Thakker, R.V.; Newey, P.J.; Walls, G.V.; Bilezikian, J.; Dralle, H.; Ebeling, P.R.; Melmed, S.; Sakurai, A.; Tonelli, F.; Brandi, M.L. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J. Clin. Endocrinol. Metab. 2012, 97, 2990–3011.
- Wu, Y.; Gao, L.; Guo, X.; Wang, Z.; Lian, W.; Deng, K.; Lu, L.; Xing, B.; Zhu, H. Pituitary adenomas in patients with multiple endocrine neoplasia type 1: A single-center experience in China. Pituitary 2019, 22, 113–123.
- Cohen-cohen, S.; Brown, D.A.; Himes, B.T.; Wheeler, L.P.; Ruff, M.W.; Major, B.T.; Ospina, N.M.S.; Atkinson, J.L.D.; Meyer, F.B.; Bancos, I.; et al. Pituitary adenomas in the setting of multiple endocrine neoplasia type 1: A single-institution experience. J. Neurosurg. 2021, 134, 1132–1138.
- Kamilaris, C.D.C.; Stratakis, C.A. Multiple endocrine neoplasia type 1 (MEN1): An update and the significance of early genetic and clinical diagnosis. Front. Endocrinol. 2019, 10, 1–7.
- Chandrasekharappa, S.C.; Guru, S.C.; Manickam, P.; Olufemi, S.E.; Collins, F.S.; Emmert-Buck, M.R.; Debelenko, L.V.; Zhuang, Z.; Lubensky, I.A.; Liotta, L.A.; et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276, 404–406.
- Concolino, P.; Costella, A.; Capoluongo, E. Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new germline variants reported in the last nine years. Cancer Genet. 2016, 209, 36–41.
- Matkar, S.; Thiel, A.; Hua, X. Menin: A scaffold protein that controls gene expression and cell signaling. Trends Biochem. Sci. 2013, 38, 394–402.
- Huang, J.; Gurung, B.; Wan, B.; Matkar, S.; Veniaminova, N.A.; Wan, K.; Merchant, J.L.; Hua, X.; Lei, M. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature 2012, 482, 542–546.
- Thakker, R.V. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol. Cell. Endocrinol. 2014, 386, 2–15.
- Pellegata, N.S.; Quintanilla-Martinez, L.; Siggelkow, H.; Samson, E.; Bink, K.; Höfler, H.; Fend, F.; Graw, J.; Atkinson, M.J. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc. Natl. Acad. Sci. USA 2006, 103, 15558–15563.
- Lee, M.; Pellegata, N.S. Multiple endocrine neoplasia type 4. Front. Horm. Res. 2013, 41, 63–78.
- Frederiksen, A.; Rossing, M.; Hermann, P.; Ejersted, C.; Thakker, R.V.; Frost, M. Clinical Features of Multiple Endocrine Neoplasia Type 4: Novel Pathogenic Variant and Review of Published Cases. J. Clin. Endocrinol. Metab. 2019, 104, 3637–3646.
- Roussel-gervais, A.; Couture, C.; Langlais, D.; Takayasu, S.; Balsalobre, A.; Rueda, B.R.; Zukerberg, L.R.; Figarella-branger, D.; Brue, T.; Drouin, J. The Cables1 Gene in Glucocorticoid Regulation of Pituitary Corticotrope Growth and Cushing Disease. J. Clin. Endocrinol. Metab. 2016, 101, 513–522.
- Matsuoka, M.; Matsuura, Y.; Semba, K.; Nishimoto, I. Molecular cloning of a cyclin-like protein associated with cyclin-dependent kinase 3 (cdk 3) in vivo. Biochem. Biophys. Res. Commun. 2000, 273, 442–447.
- Hernández-ramírez, L.C.; Gam, R.; Valdés, N.; Lodish, M.B.; Pankratz, N.; Balsalobre, A.; Gauthier, Y.; Faucz, F.R. Loss-of-function mutations in the CABLES1 gene are a novel cause of Cushing ’ s disease. Endocr.-Relat. Cancer 2017, 24, 379–392.
- Zukerberg, L.R.; Patrick, G.N.; Nikolic, M.; Humbert, S.; Wu, C.L.; Lanier, L.M.; Gertler, F.B.; Vidal, M.; Van Etten, R.A.; Tsai, L.H. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron 2000, 26, 633–646.
- Alrezk, R.; Suarez, A.; Tena, I.; Pacak, K. Update of Pheochromocytoma Syndromes: Genetics, Biochemical Evaluation, and Imaging. Front. Endocrinol. 2018, 9, 515.
- Xekouki, P.; Pacak, K.; Almeida, M.; Wassif, C.A.; Rustin, P.; Nesterova, M.; De La Luz Sierra, M.; Matro, J.; Ball, E.; Azevedo, M.; et al. Succinate dehydrogenase (SDH) D subunit (SDHD) inactivation in a growth-hormone-producing pituitary tumor: A new association for SDH? J. Clin. Endocrinol. Metab. 2012, 97, 357–366.
- Xekouki, P.; Szarek, E.; Bullova, P.; Giubellino, A.; Quezado, M.; Mastroyannis, S.A.; Mastorakos, P.; Wassif, C.A.; Raygada, M.; Rentia, N.; et al. Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in humans and mice. J. Clin. Endocrinol. Metab. 2015, 100, E710–E719.
- Papathomas, T.G.; Gaal, J.; Corssmit, E.P.M.; Oudijk, L.; Korpershoek, E.; Heimdal, K.; Bayley, J.P.; Morreau, H.; Van Dooren, M.; Papaspyrou, K.; et al. Non-pheochromocytoma (PCC)/paraganglioma (PGL) tumors in patients with succinate dehydrogenase-related PCC-PGL syndromes: A clinicopathological and molecular analysis. Eur. J. Endocrinol. 2014, 170, 1–12.
- Moosavi, B.; Zhu, X.L.; Yang, W.C.; Yang, G.F. Molecular pathogenesis of tumorigenesis caused by succinate dehydrogenase defect. Eur. J. Cell Biol. 2020, 99, 151057.
- Moosavi, B.; Berry, E.A.; Zhu, X.L.; Yang, W.C.; Yang, G.F. The assembly of succinate dehydrogenase: A key enzyme in bioenergetics. Cell. Mol. Life Sci. 2019, 76, 4023–4042.
- Xekouki, P.; Brennand, A.; Whitelaw, B.; Pacak, K.; Stratakis, C.A. The 3PAs: An Update on the Association of Pheochromocytomas, Paragangliomas, and Pituitary Tumors. Horm. Metab. Res. 2019, 51, 419–436.
- Dénes, J.; Swords, F.; Rattenberry, E.; Stals, K.; Owens, M.; Cranston, T.; Xekouki, P.; Moran, L.; Kumar, A.; Wassif, C.; et al. Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: Results from a large patient cohort. J. Clin. Endocrinol. Metab. 2015, 100, E531–E541.
- Foulkes, W.D.; Priest, J.R.; Duchaine, T.F. DICER1: Mutations, microRNAs and mechanisms. Nat. Rev. Cancer 2014, 14, 662–672.
- Song, M.S.; Rossi, J.J. Molecular mechanisms of Dicer: Endonuclease and enzymatic activity. Biochem. J. 2017, 474, 1603–1618.
- Faure, A.; Atkinson, J.; Bouty, A.; Brien, M.O. DICER1 pleuropulmonary blastoma familial tumour predisposition syndrome: What the paediatric urologist needs to know. J. Pediatr. Urol. 2016, 12, 5–10.
- Sahakitrungruang, T.; Srichomthong, C.; Pornkunwilai, S.; Amornfa, J.; Shuangshoti, S.; Kulawonganunchai, S.; Suphapeetiporn, K.; Shotelersuk, V. Germline and Somatic DICER1 Mutations in a Pituitary Blastoma Causing Infantile-Onset Cushing’s Disease. J. Clin. Endocrinol. Metab. 2014, 99, E1487–E1492.
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134.
- Manoranjan, B.; Mahendram, S.; Almenawer, S.A.; Venugopal, C.; McFarlane, N.; Hallett, R.; Vijayakumar, T.; Algird, A.; Murty, N.K.; Sommer, D.D.; et al. The identification of human pituitary adenoma-initiating cells. Acta Neuropathol. Commun. 2016, 4, 125.
- Würth, R.; Barbieri, F.; Pattarozzi, A.; Gaudenzi, G.; Gatto, F.; Fiaschi, P.; Ravetti, J.L.; Zona, G.; Daga, A.; Persani, L.; et al. Phenotypical and Pharmacological Characterization of Stem-Like Cells in Human Pituitary Adenomas. Mol. Neurobiol. 2017, 54, 4879–4895.
- Peverelli, E.; Giardino, E.; Treppiedi, D.; Meregalli, M.; Belicchi, M.; Vaira, V.; Corbetta, S.; Verdelli, C.; Verrua, E.; Serban, A.L.; et al. Dopamine receptor type 2 (DRD2) and somatostatin receptor type 2 (SSTR2) agonists are effective in inhibiting proliferation of progenitor/stem-like cells isolated from nonfunctioning pituitary tumors. Int. J. Cancer 2017, 140, 1870–1880.
- Mertens, F.; Gremeaux, L.; Chen, J.; Fu, Q.; Willems, C.; Roose, H.; Govaere, O.; Roskams, T.; Cristina, C.; Becú-Villalobos, D.; et al. Pituitary tumors contain a side population with tumor stem cell-associated characteristics. Endocr. Relat. Cancer 2015, 22, 481–504.
- Xu, Q.; Yuan, X.; Tunici, P.; Liu, G.; Fan, X.; Xu, M.; Hu, J.; Hwang, J.Y.; Farkas, D.L.; Black, K.L.; et al. Isolation of tumour stem-like cells from benign tumours. Br. J. Cancer 2009, 101, 303–311.
- Chen, L.; Ye, H.; Wang, X.; Tang, X.; Mao, Y.; Zhao, Y.; Wu, Z.; Mao, X.O.; Xie, L.; Jin, K.; et al. Evidence of brain tumor stem progenitor-like cells with low proliferative capacity in human benign pituitary adenoma. Cancer Lett. 2014, 349, 61–66.
- Wierinckx, A.; Roche, M.; Legras-lachuer, C. MicroRNAs in pituitary tumors. Mol. Cell. Endocrinol. 2017, 456, 51–61.
- Bottoni, A.; Piccin, D.; Tagliati, F.; Luchin, A.; Zatelli, M.C.; Uberti, E.C.D. miR-15a and miR-16-1 down-regulation in pituitary adenomas. J. Cell. Phys. 2005, 204, 280–285.
- Butz, H. Circulating Noncoding RNAs in Pituitary Neuroendocrine Tumors—Two Sides of the Same Coin. Int. J. Mol. Sci. 2022, 23, 5122.
- Xu, D.; Wang, L. The Involvement of miRNAs in Pituitary Adenomas Pathogenesis and the Clinical Implications. Eur. Neurol. 2022, 85, 171–176.
- Butz, H.; Patócs, A. MicroRNAs in endocrine tumors. EJIFCC 2019, 30, 146.
- Zhang, R.; Yang, F.; Fan, H.; Wang, H.; Wang, Q.; Yang, J.; Song, T. Long non-coding RNA TUG1/microRNA-187-3p/TESC axis modulates progression of pituitary adenoma via regulating the NF- κ B signaling pathway. Cell Death Dis. 2021, 12, 524.
- Zhou, K.; Zhang, T.; Fan, Y.; Du, G.; Wu, P. MicroRNA-106b promotes pituitary tumor cell proliferation and invasion through PI3K/AKT signaling pathway by targeting PTEN. Tumor Biol. 2016, 37, 13469–13477.
- Renjie, W.; Haiqian, L. MiR-132, miR-15a and miR-16 synergistically inhibit pituitary tumor cell proliferation, invasion and migration by targeting Sox5. Cancer Lett. 2015, 356, 568–578.