Type 2 diabetes mellitus (DM) represents, with its macro and microvascular complications, one of the most critical healthcare issues. The pathogenesis of DM shares with cardiovascular diseases (CAD) a complex landscape of risk factors, including genetic predisposition and various environmental factors like a high-fat diet, sedentary lifestyle, and chronic stress. In particular, CAD is the leading cause of morbidity and mortality in diabetic patients, determining a significant impact on life expectancy. Notably, DM is equivalent to established ischemic CAD risk, and patients with diabetes have a two- to four-fold greater risk of developing CAD than non-diabetic patients. Myocardial infarction, ischemic ictus, and peripheral arterial disease are the main expression of DM progression and, often, the first event in diabetic patients. CAD and DM strictly depend on various inflammatory pathways that are able to promote the onset and development of insulin resistance, atherosclerotic plaque, and heart failure (HF).
- type 2 diabetes mellitus
- meta-inflammation
- cardiovascular diseases
1. Meta-Inflammation and Metabolic Endotoxemia
2. Meta-Inflammation and the “Unfolded Protein Response” (UPR)
3. The Innate Immune Response
4. The Adaptive Immune Response
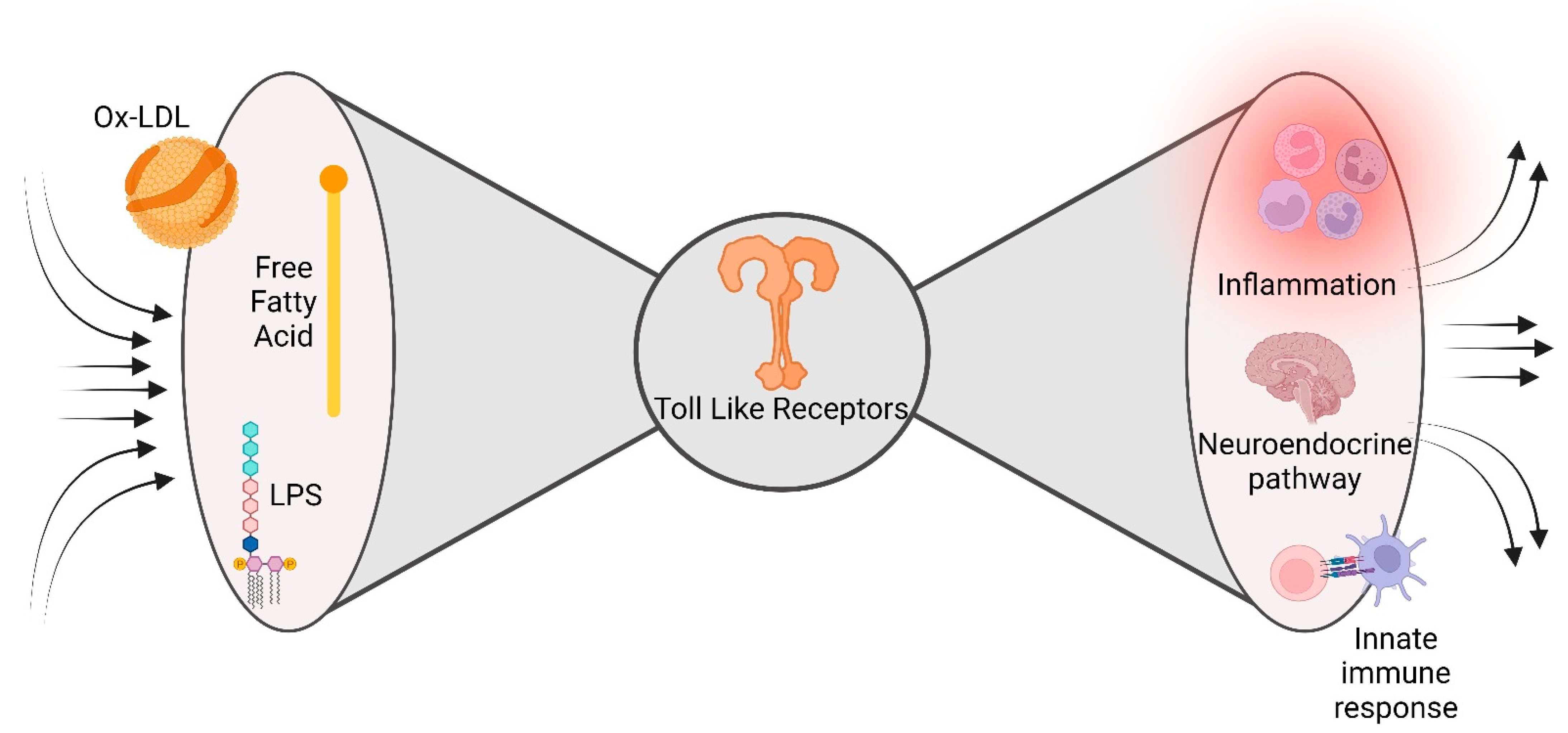
5. The Bow Tie Model
References
- Erridge, C.; Attina, T.; Spickett, C.M.; Webb, D.J. A high-fat meal induces low-grade endotoxemia: Evidence of a novel mechanism of postprandial inflammation. Am. J. Clin. Nutr. 2007, 86, 1286–1292.
- Pahwa, R.; Devaraj, S.; Jialal, I. The effect of the accessory proteins, soluble CD14 and lipopolysaccharide-binding protein on Toll-like receptor 4 activity in human monocytes and adipocytes. Int. J. Obes. 2016, 40, 907–911.
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188.
- Biasucci, L.M.; La Rosa, G.; Pedicino, D.; D’aiello, A.; Galli, M.; Liuzzo, G. Where Does Inflammation Fit? Curr. Cardiol. Rep. 2017, 19, 84.
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated Circulating Levels of Tumor Necrosis Factor in Severe Chronic Heart Failure. N. Engl. J. Med. 1990, 323, 236–241.
- Kunkel, S.L.; Spengler, M.; May, M.A.; Spengler, R.; Larrick, J.; Remick, D. Prostaglandin E2 regulates macrophage-derived tumor necrosis factor gene expression. J. Biol. Chem. 1988, 263, 5380–5384.
- Giordano, A.; Murano, I.; Mondini, E.; Perugini, J.; Smorlesi, A.; Severi, I.; Barazzoni, R.; Scherer, P.E.; Cinti, S. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J. Lipid Res. 2013, 54, 2423–2436.
- Gregor, M.F.; Hotamisligil, G.S. Thematic review series: Adipocyte Biology. Adipocyte stress: The endoplasmic reticulum and metabolic disease. J. Lipid Res. 2007, 48, 1905–1914.
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529.
- Todd, D.J.; Lee, A.-H.; Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008, 8, 663–674.
- Hu, P.; Han, Z.; Couvillon, A.D.; Kaufman, R.J.; Exton, J.H. Autocrine Tumor Necrosis Factor Alpha Links Endoplasmic Reticulum Stress to the Membrane Death Receptor Pathway through IRE1α-Mediated NF-κB Activation and Down-Regulation of TRAF2 Expression. Mol. Cell. Biol. 2006, 26, 3071–3084.
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic Reticulum Stress Activates Cleavage of CREBH to Induce a Systemic Inflammatory Response. Cell 2006, 124, 587–599.
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337.
- Pedicino, D.; Giglio, A.F.; Galiffa, V.A.; Cialdella, P.; Trotta, F.; Graziani, F.; Liuzzo, G. Infections, immunity and atherosclerosis: Pathogenic mechanisms and unsolved questions. Int. J. Cardiol. 2013, 166, 572–583.
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23.
- Shioi, T.; Matsumori, A.; Kihara, Y.; Inoko, M.; Ono, K.; Iwanaga, Y.; Yamada, T.; Iwasaki, A.; Matsushima, K.; Sasayama, S. Increased Expression of Interleukin-1β and Monocyte Chemotactic and Activating Factor/Monocyte Chemoattractant Protein-1 in the Hypertrophied and Failing Heart With Pressure Overload. Circ. Res. 1997, 81, 664–671.
- Pedicino, D.; Severino, A.; Ucci, S.; Bugli, F.; Flego, D.; Giglio, A.F.; Trotta, F.; Ruggio, A.; Lucci, C.; Iaconelli, A.; et al. Epicardial adipose tissue microbial colonization and inflammasome activation in acute coronary syndrome. Int. J. Cardiol. 2017, 236, 95–99.
- Liuzzo, G.; Montone, R.A.; Gabriele, M.; Pedicino, D.; Giglio, A.F.; Trotta, F.; Galiffa, V.A.; Previtero, M.; Severino, A.; Biasucci, L.M.; et al. Identification of unique adaptive immune system signature in acute coronary syndromes. Int. J. Cardiol. 2013, 168, 564–567.
- Kintscher, U.; Hartge, M.; Hess, K.; Foryst-Ludwig, A.; Clemenz, M.; Wabitsch, M.; Fischer-Posovszky, P.; Barth, T.F.; Dragun, D.; Skurk, T.; et al. T-lymphocyte infiltration in visceral adipose tissue: A primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arter. Thromb. Vasc. Biol. 2008, 28, 1304–1310.
- Madhumitha, H.; Mohan, V.; Deepa, M.; Babu, S.; Aravindhan, V. Increased Th1 and suppressed Th2 serum cytokine levels in subjects with diabetic coronary artery disease. Cardiovasc. Diabetol. 2014, 13, 1.
- McGillicuddy, F.C.; Chiquoine, E.H.; Hinkle, C.C.; Kim, R.J.; Shah, R.; Roche, H.M.; Smyth, E.M.; Reilly, M.P. Interferon γ Attenuates Insulin Signaling, Lipid Storage, and Differentiation in Human Adipocytes via Activation of the JAK/STAT Pathway. J. Biol. Chem. 2009, 284, 31936–31944.
- Ruggio, A.; Pedicino, D.; Flego, D.; Vergallo, R.; Severino, A.; Lucci, C.; Niccoli, G.; Trani, C.; Burzotta, F.; Aurigemma, C.; et al. Correlation between CD4+CD28null T lymphocytes, regulatory T cells and plaque rupture: An Optical Coherence Tomography study in Acute Coronary Syndromes. Int. J. Cardiol. 2019, 276, 289–292.
- Canonico, F.; Pedicino, D.; Severino, A.; Vinci, R.; Flego, D.; Pisano, E.; D’aiello, A.; Ciampi, P.; Ponzo, M.; Bonanni, A.; et al. GLUT-1/PKM2 loop dysregulation in patients with non-ST-segment elevation myocardial infarction promotes metainflammation. Cardiovasc. Res. 2022; Online ahead of print.
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Patel, B.; Hamid, T.; Rokosh, G.; Prabhu, S.D. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ. Heart Fail. 2017, 10, e003688.
- Tieri, P.; Grignolio, A.; Zaikin, A.; Mishto, M.; Remondini, D.; Castellani, G.C.; Franceschi, C. Network, degeneracy and bow tie. Integrating paradigms and architectures to grasp the complexity of the immune system. Theor. Biol. Med. Model. 2010, 7, 32.
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590.
- Lee, J.Y.; Zhao, L.; Youn, H.S.; Weatherill, A.R.; Tapping, R.; Feng, L.; Lee, W.H.; Fitzgerald, K.A.; Hwang, D.H. Saturated Fatty Acid Activates but Polyunsaturated Fatty Acid Inhibits Toll-like Receptor 2 Dimerized with Toll-like Receptor 6 or 1. J. Biol. Chem. 2004, 279, 16971–16979.
- Schiattarella, G.G.; Rodolico, D.; A Hill, J. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovasc. Res. 2021, 117, 423–434.