2. Adenosine-Pathway-Mediated Immunosuppression
Adenosine triphosphate (ATP) is the universal cellular energy currency and is present throughout the extracellular environment. Under normal physiological conditions, the extracellular levels of ATP are tightly regulated to remain in the nanomolar range. However, during inflammation, ischemia/hypoxia, tissue injury, or tumorigenesis extracellular ATP levels can increase to the micromolar range due to its release from inflammatory, apoptotic, or necrotic cells
[6][9]. Extracellular ATP signals through P2 purinergic receptors (P2Rs) that are broadly expressed by both immune and non-immune cells and drives multiple physiological and pathological processes
[7][10]. The current model of purinergic signaling effects on the immune response describes a delicate balance between the pro-inflammatory “danger-signal” activity of ATP and the anti-inflammatory, cytoprotective, and immunosuppressive functions of extracellular adenosine (eADO)
[8][11]. ATP is a major regulator of inflammatory signaling and can act as a damage-associated molecular pattern (DAMP). As a DAMP, ATP can activate the NLRP3 inflammasome in cells by binding purinergic P2X7 receptors
[9][12]. Inflammasomes are large protein complexes that play a crucial role in the innate immune system by sensing pathogenic or exogenous danger signals and triggering an inflammatory response through downstream caspase signaling cascades
[10][13]. Of the canonical inflammasomes, NLRP3 has been found to play a key role in the TME in mediating tumor-promoting inflammation, immune evasion, proliferative signaling, invasion, angiogenesis, and inhibition of apoptosis
[11][12][14,15]. Additional ATP released from stressed and/or dying cells can also induce immunogenic cell death and is important for immune cell recognition of active infection, while accumulation of adenosine aims to restore tissue homeostasis and prevent an excessive inflammatory reaction. Like ATP, eADO in normal tissues is held constant around nanomolar concentrations but can increase over 100-fold in the TME
[13][16]. Under normal physiological conditions, adenosine is mainly found intracellularly and is generated during the conversion of S-adenosylhomocysteine to adenosine and homocysteine by Adenosylhomocysteinase
[14][17]. The amount of adenosine in the extracellular space is controlled by equilibrative nucleoside transporters (ENTs), which shuttle adenosine based on its concentration gradient, and concentrative nucleoside transporters (CNTs), which allow the intracellular influx of adenosine against its concentration gradient. In healthy tissues, adenosine regulates numerous physiological processes but mainly acts to maintain homeostasis following injury by restricting immune responses and promoting wound healing
[15][18]. In contrast, the levels of eADO in the TME are regulated by multiple systems of enzymes and transporters that produce, degrade, and recycle purine metabolites (
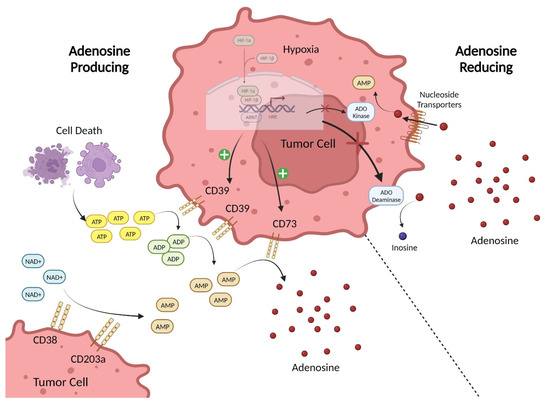
Figure 1). Extracellular adenosine in the TME can be produced by simple diffusion or active transport of intracellular adenosine but is mainly generated through two pathways. In the first, ATP in the extracellular environment is hydrolyzed to ADP and AMP, and then AMP is converted to eADO by the ectonucleotidases CD39 and CD73
[16][19]. Therefore, eADO can rapidly accumulate in the TME due to the higher levels of ATP present and the overexpression of CD39 and CD73 found on multiple cell types in the TME
[17][20]. In the second pathway, AMP is produced from NAD+ by the enzymatic actions of CD38 and CD203a, and is then converted to eADO by CD73
[18][21]. Furthermore, cancer cells commonly have genetic mutations that promote altered purine metabolism to facilitate increased production or reduced degradation of eADO
[19][22]. Extracellular adenosine is removed from the environment by cell surface ADO deaminase, which converts it to inosine, and by nucleoside transporters that bring eADO intracellularly for conversion back to AMP by ADO kinases
[20][23]. Hypoxic conditions, such as those found in the TME, can inhibit these eADO-consuming pathways, which further amplifies the increase in adenosine signaling
[21][24].
Figure 1. Sources of Adenosine in the TME. The major adenosine (eADO)-producing pathway in the TME involves the processing of the precursor ATP by ectonucleotidases. ATP is released into the extracellular environment due to cell death. ATP accumulating in the extracellular milieu is then enzymatically converted via the canonical pathway involving the sequential hydrolysis of ATP to ADP and AMP by CD39 and subsequently the hydrolysis of AMP to eADO by CD73. In the non-canonical pathway, precursor NAD+ substrate is acted on by CD38 to generate ADP-ribose (ADPR) that can be converted to AMP by CD203a, which is then hydrolyzed by CD73 to produce eADO. To reduce eADO levels in the extracellular space, adenosine can be re-uptaken into the cell via nucleoside transporters, where it is converted to AMP by ADO kinase or converted to inosine by cell surface ADO deaminase. Hypoxic signaling in tumor cells promotes expression of CD39 and CD73 while simultaneously inhibiting ADO kinase and ADO deaminase activity. Created with BioRender.com.
The main signaling actions of eADO are mediated by four G-protein-coupled receptors classified as the subtypes A1, A2
A, A2
B, and A3. The A1, A2
A, and A3 subtypes are considered high-affinity, and A2
B by contrast is a low-affinity receptor that is only active in pathological conditions when eADO levels are elevated
[22][25]. Adenosine receptors are widely distributed in many different tissues including the nervous, cardiovascular, gastrointestinal, and immune systems due to adenosine’s role as a ubiquitous extracellular signaling molecule
[23][26]. In addition to regulating metabolic homeostasis in most cells by manipulating adenosine levels, adenosine receptors play important roles in modulating cardiac contractility and vasodilation, synaptic transmission in the brain and sleep cycles, and inflammatory responses
[24][27]. The A1 and A3 subtypes are coupled to G
i/o and inhibit the activity of adenylate cyclase, whereas A2
A and A2
B receptors are G
s family members and activate adenylate cyclase, thus triggering cAMP-dependent downstream signaling events. Additionally, adenosine receptors can signal through cAMP-independent pathways. The A2
B and A3 receptors can trigger phospholipase C-mediated pathway activation via G
q/11 proteins and A2
A and A2
B receptors have been found to activate ERK, p38 MAPK, and/or PI3K–AKT–mTOR-dependent pathways in various cell types
[25][28]. In an oncogenic setting, the immunosuppressive effects of eADO are mainly induced via binding by the A2
A and A2
B receptors
[26][29]. These two receptor subtypes are present on a variety of cell types located in the TME, with A2
A receptors ubiquitously expressed by immune cells and A2
B receptors predominantly found on myeloid cells. The adenosine receptor system can exert a variety of effects on the differentiation, maturation, and activation state of cells in the TME depending on cell type (
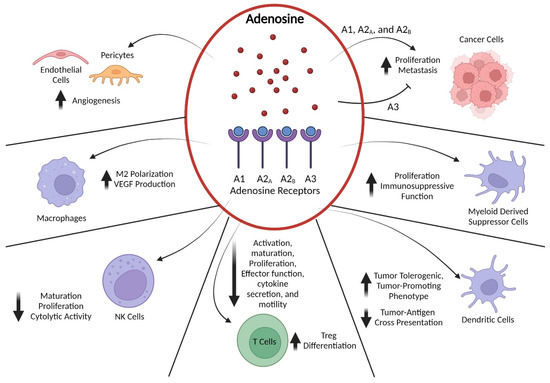
Figure 2).
Figure 2. Adenosine Signaling Effects on Cells in the TME. Adenosine accumulated in the TME exerts its effects on a variety of cell types by binding to A1, A2A, A2B, and A3 receptors. Signaling through A2A and A2B receptors induces cyclic AMP (cAMP)-dependent pathways, whereas activation of A1 and A3 receptors inhibits cAMP generation. In cancer cells, adenosine binding to A1, A2A, and A2B receptors stimulates proliferation, invasion, and metastasis, whereas binding to A3 receptors inhibits those processes. Both A2A and A2B receptor signaling exert potent immunomodulatory effects on myeloid-derived suppressor cells and enhance their immunosuppressive cytokine secretion. Similarly, A2A and A2B receptor signaling suppresses professional antigen-presenting cells such as dendritic cells by reducing their capacity for tumor-antigen cross-presentation through downregulation of MHC class II expression, while also promoting their production of tumor-tolerogenic and/or -promoting factors such as IL-6, IL-10, and TGFβ. In T cells, A2A binding leads to a multi-layered suppression of anti-tumor immune function by dampening T-cell receptor and co-stimulatory signals, thereby blocking effector T-cell activation, maturation, proliferation, motility, and secretion of multiple cytokines. Additionally, activation of A2A receptors in CD4+ T cells promotes their differentiation into regulatory T (Treg) cells and increases their immunosuppressive capabilities. Likewise, NK cells express high levels of A2A receptors that, when bound by adenosine, limit their maturation, proliferation, and cytotoxic function. Activation of A2A and A2B receptors in macrophages triggers differentiation into the tumor-associated M2 phenotype that produces tumor-promoting cytokines such as vascular endothelial growth factor (VEGF). Finally, adenosine-mediated signaling through A2A and A2B receptors on endothelial cells and pericytes promotes angiogenesis and limits trafficking of immune cells. Created with BioRender.com.
3. Preclinical and Clinical Data Supporting Adenosine-Pathway-Targeted Therapies
Cancer immunotherapies such as immune checkpoint blockade (ICB) have revolutionized cancer treatment and have renewed interest in therapies that can restore anti-tumor immunity. However, despite the notable successes of ICB, only a minority of solid tumor patients have durable responses, which has highlighted the need for further research in understanding immune evasion mechanisms to identify novel therapeutic targets
[27][43]. As described in the previous sections, the adenosine signaling pathway has been shown to regulate immune cell responses in the TME and is an attractive target for immunotherapeutic development. There is a wealth of both preclinical and clinical data that support further interrogation of adenosine signaling inhibition for numerous solid tumors. Adenosine signaling pathway components such as CD39, CD73, and A2
A receptors are overexpressed by multiple cell types in the TME of various cancers, including colorectal, gastric, head and neck, breast, and brain cancer, and often correlate with an immunosuppressive signature, aggressiveness, and poor prognosis in patients
[28][29][30][31][32][44,45,46,47,48]. In 1975, Chu et al. became the first to demonstrate that eADO could suppress T-cell activity against cancer cells in vitro
[33][49]. Several landmark studies subsequently established the primary role of A2
A receptors in mediating immunosuppressive signaling and that targeting adenosine receptors could potently enhance anti-tumor responses both in vitro and in vivo
[34][35][36][50,51,52]. Additional preclinical evidence has shown that targeting the production of eADO in the TME via CD39 and CD73 is another promising strategy for restoring anti-tumor immunity. Reducing CD73 expression in tumor cell lines sensitized them to T-cell-mediated killing, and anti-CD73 antibodies could reduce tumor growth and metastasis by activating NK and T-cell responses
[37][38][53,54]. Likewise, CD39-targeting monoclonal antibodies have been shown to inhibit eADO production in the TME, effectively suppress metastasis, upregulate the expression of activation markers and cytotoxic granule components in immune cells, and increase the anti-tumor functions of infiltrating NK and T cells
[39][40][41][55,56,57]. Monoclonal antibodies and small-molecule inhibitors that target CD39, CD73, and adenosine receptors are all under development for cancer therapy. Compared to anti-CD39 and anti-CD73 antibodies, small-molecule inhibitors may have better penetration into solid tumors and become more widely available in the TME
[42][58]. Given the accumulating preclinical research indicating the benefit of targeting eADO to increase anti-tumor immunity, there has been accelerated initiation of clinical trials.
Early results from clinical trials of eADO-pathway-targeting agents have demonstrated good tolerability and some efficacy as a monotherapy. One of the first drugs developed to target adenosine-mediated immunosuppression was the dual A2
A/A2
B receptor antagonist AB928 or etrumadenant, which was found to be safe, orally bioavailable, and possessed immunomodulatory activity in healthy volunteers
[43][59]. Etrumadenant is now being further evaluated in multiple phase I/II studies for its anti-cancer efficacy
[44][60]. The first clinical report confirming the utility of adenosine pathway antagonism for cancer treatment came from a phase I trial of CPI-444 or ciforadenant, a small-molecule inhibitor with selectivity for A2
A receptors, which demonstrated monotherapy activity in renal cell carcinoma patients, even those who were treatment-refractory to anti-PD-L1 therapy
[45][61]. Phase I studies of additional A2
A receptor antagonists further established these drugs as well tolerated with minimal side effects and capable of inducing clinical responses as both a monotherapy and in combination with other immunotherapies
[46][47][62,63]. Several anti-CD39 and anti-CD73 monoclonal antibodies have also begun entering clinical trials.
4. Potential Combinations with Adenosine-Pathway-Targeted Therapy
Strong rationale exists for combining adenosine-pathway-targeted therapies with traditional chemotherapy or immunotherapy. Immune checkpoint blockade can activate T cells and increase their infiltration into solid tumors, but resistance is common. Adenosine signaling is a non-redundant immunosuppressive mechanism in the TME that, if inhibited, could further unleash the cytotoxic potential of infiltrating immune cells. First, there are multiple pathways that contribute to eADO-mediated immunosuppression in the TME; therefore, there may be synergy when combining eADO-generating pathway inhibition with eADO receptor antagonism. A study of dual therapy with an anti-CD73 mAb and an A2
A receptor antagonist found there was superior anti-tumor immunity with the combination compared to either treatment alone
[48][69]. Early reports from a clinical trial investigating a combination of A2
A receptors and CD73 inhibition similarly found an increase in response rate without a marked increase in the adverse event rate compared to either monotherapy
[49][70]. Another potential strategy for enhancing efficacy is the co-inhibition of CD39 and CD73, and this is supported by preclinical data indicating compensatory mechanisms when either is targeted alone
[50][71]. Moreover, CD39 and CD73 have pro-tumorigenic functions beyond their enzyme activity, including potentiating tumor cell adhesion, migration, and metastasis
[51][72]. For example, an antibody targeting CD73 that did not affect eADO production could induce internalization of CD73, which inhibited metastasis formation
[52][73]. Thus, combining adenosine-pathway-targeted agents with other therapies may have further additive or synergistic effects.