Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Minakshi Saikia and Version 2 by Lindsay Dong.
Molecules involved in drug resistance can be targeted for better therapeutic efficacies. Research on midkine (MDK) has escalated in the last few decades, which affirms a positive correlation between disease progression and MDK expression in most cancers and indicates its association with multi-drug resistance in cancer. MDK, a secretory cytokine found in blood, can be exploited as a potent biomarker for the non-invasive detection of drug resistance expressed in various cancers and, thereby, can be targeted.
- midkine
- drug resistance
- cancer targeting
- transcriptional regulators
1. Introduction
Midkine (MDK), encoded by the MDK gene, was first discovered through differential hybridization by examining the increased RNA levels of its cDNA in retinoic acid-induced differentiation in embryonal carcinoma cells [1]. This 13 kDa cysteine-rich protein consists of two main domains, each containing three antiparallel β-strands and multiple heparin-binding consensus sites, which, when bound to, prompt chemical and structural changes within the protein [2]. MDK is thus categorized as a heparin-binding protein, belonging to the family of heparin-binding growth associate molecules (HB-GAM), which contains one other protein 50% similar to MDK, pleiotrophin (PTN), both of which share identical functions [3].
MDK plays an essential role in developing and maintaining crucial systems, such as the nervous system. It is also known as a neurite growth-promoting factor as it helps in the development and survival of neurons [4]. Formerly, it was identified as developmentally vital as a retinoic acid-responsive gene product during mid-gestation, and hence it was named Mid-kine. Recent studies have highlighted the role of MDK as a biomarker due to its abnormally high expression in various malignancies, unlike in normal tissues with a weak or minimum expression. MDK mediates cell growth, survival, metastasis, and angiogenesis and accomplishes all the major hallmarks of cancer [5] (Figure 1).
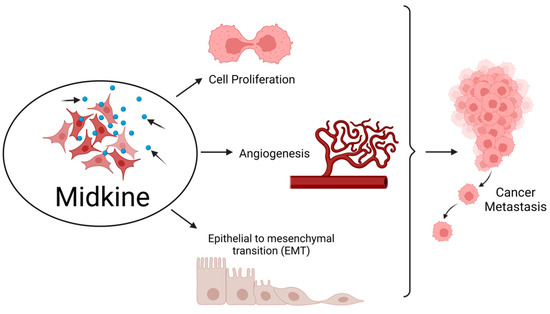
Figure 1. Midkine is involved in the hallmarks of cancer. MDK enhances cell proliferation and angiogenesis and also epithelial to mesenchymal transition (EMT), thereby aiding in metastasis of tumor from the primary site to distant locations. Created with BioRenderBioRender.com.com.
MDK is a soluble growth factor secreted by the cells that produce it [6]. In the case of cancer, multiple sclerosis, ischemia, and other inflammation and neural diseases, MDK is a cytokine responsible for survival and proliferation in all of these conditions [7][8][9][10][7,8,9,10]. The presence of MDK in serum is associated with worse outcomes. When MDK is genetically silenced, it leads to a decrease in the proliferation of cancer cells [11]. While the exact pathways are not entirely known, the presence and association of MDK with carcinogenesis is evident [12].
MDK is overexpressed in at least 20 different cancers compared to normal levels in healthy individuals [6]. In pancreatic cancer cells, there was an increase in proliferation when MDK-depleted cells were exposed to MDK compared to the MDK-depleted cells without the treatment [13].
MDK overexpression can be correlated with the diagnosis and disease prognosis in various cancers, and it can be a promising molecular target in personalized medicine. Since it is a secretory cytokine and can be easily detected in body fluids such as blood, cerebrospinal fluid, urine, or tumor mRNA analysis, it is an inexpensive and convenient biomarker for the early diagnosis and prognosis, and also monitoring of the treatment response for NSCLC [6][14][6,14]. MDK is part of the diagnostic biomarker blood test that can detect early-stage lung cancer in at-risk populations [6].
2. MDK Drives Oncogenesis via Various Pathways
MDK is found to drive oncogenesis by various pathways (Figure 2).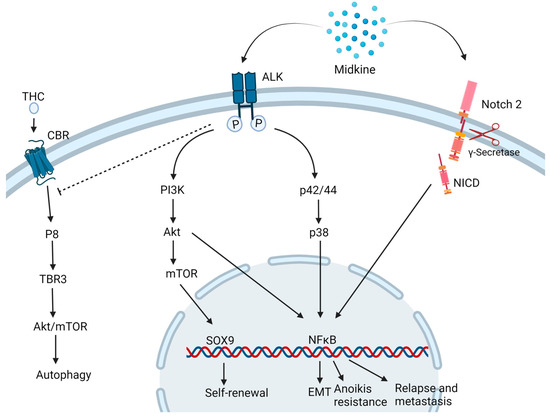
Figure 2. Midkine drives oncogenesis via different pathways. MDK binding to ALK receptor inhibits the autophagic cell death induced by Tetrahydrocannabinol (THC) and activates the PI3K/Akt/mTOR pathways inducing to self-renewal and stemness properties to the cells. Also, the MAPK pathway activates NFκB which triggers multiple downstream mechanisms of carcinogenesis such as epithelial to mesenchymal transition (EMT), Anoikis resistance and relapse and metastasis. MDK also binds to Notch 2 followed by the cleavage of Notch intracellular domain (NICD) by γ-secretase and activation of NFκB. CBR: Cannabinoid receptor. Created with BioRender.com.
In a neuroblastoma and osteosarcoma cells study, MDK induced cytoprotective function in the neighboring drug-sensitive cells against cell death induced by doxorubicin via the Akt pathway [15][19]. Additionally, in neuronal cell line PC12, MDK is found to impart neuroprotective functions via ERK activation [16][28]. In prostate cancer, MDK was found to be induced by TNF-α via the NFκB pathway. It activates the extracellular signal-regulated kinase ½ (Erk1/2) and p38 mitogen-activated protein kinase (MAPK) pathways in prostate cancer and LNCaP cells [17][29]. This cytokine is also linked to inducing EMT-mediated NF-κB activation upon interaction with the Notch 2 receptor [18][30] and angiogenesis, hence playing a key role in metastasis. MDK-Notch 2 signaling has also been shown to promote neuroblastoma through the downstream effector Hes-1 [19][31]. The binding of MDK to the anaplastic lymphoma kinase (ALK) receptors impedes the autophagic cell death-inducing activity of tetrahydrocannabinol (THC) which is the active component of marijuana, by preventing the upregulation of endoplasmic reticulum stress-related protein P8 and TRB3 [20][32]. MDK/ALK receptor axis is also noted to maintain the self-renewal capacity of the glioma-initiating cells (GICs) by preventing the autophagic degradation of the transcription factor SOX9 [21][33]. MDK activates mTOR by phosphorylating mTOR target RPS6 in uveal melanoma cells [22][34].
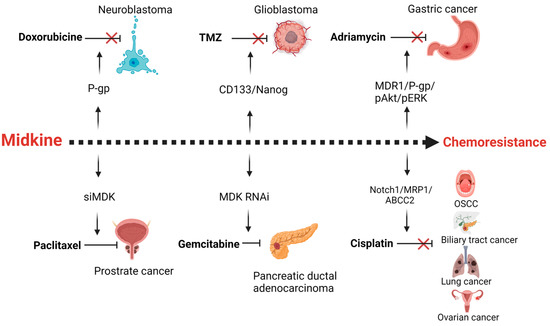
3. MDK and Drug Resistance
Drug resistance is the major obstacle in successful treatment of cancer. Resistance can be due to inherent mechanisms or acquired due to cancer cell adaptation to the chemotherapy stress. These mechanisms include an increase in efflux of the chemotherapeutic agents through the overexpression of drug transport proteins or activation of the DNA repair mechanisms, alterations in the drug target interactions, or alterations in the apoptotic response. MDK is found to be a putative gene involved in the drug resistance of various cancers (Figure 3).Figure 3. Midkine is a mediator of resistance of different chemotherapeutic agents in various cancers. MDK up-regulates p-glycoprotein (P-gp) in neuroblastoma cells in response to Doxorubicine chemotherapy. The Cancer stemness of the glioblastoma is enhanced by midkine upon temozolomide treatment. In gastric cancer adriamycin therapy results in the enhanced expression of MDK mediated multidrug resistance proteins. Cisplatin therapy fails in oral squamous cell carcinoma, biliary tract cancer, lung and ovarian cancer due to the expression of drug efflux pumps, mediated by MDK. Inhibition of MDK by siRNA or RNAi leads to increased sensitivity of paclitaxel and gemcitabine in prostrate and pancreatic ductal adenocarcinoma. Created with BioRenderBioRender.com.com.
One study completed in ESCC (esophageal squamous cell carcinoma) found that out of 66 ESCC samples studied using immunohistochemistry, MDK was over-expressed with a positive rate of 56.1% (37/66) [23][35]. Another group has shown the direct association of high expression of MDK with adverse OS in solid tumor patients by performing a meta-analysis on a total of 2097 patients from 17 observational studies [24][36]. Hu et al. have demonstrated in patient samples that intracellular Rhodamine123 accumulation in B cell lineage Acute lymphoblastic leukemia (ALL) cells is much lower than in normal cells, which positively correlates with the high MDK gene expression in those cells. They suggested that MDK might regulate drug efflux pumps [25][24]. In Pancreatic ductal adenocarcinoma (PDAC), as the dose of gemcitabine increased, so did the amount of MDK. When cells were depleted of MDK, they were no longer resistant to gemcitabine [18][30]. In melanomas, MDK effectively trains macrophages and cytotoxic T cells to prevent the immune system from attacking and recognizing the cancer cells [26][37]. Similarly, in a gastric cancer cell line, when MDK was silenced, there was a decrease in cell survival, indicating that a reduction of MDK might be involved in chemosensitization. Similarly, in HeLa cells, when MDK was overexpressed, there was a decrease in sensitivity to chemotherapeutic agents [27][38].
When human meningioma cells were treated with camptothecin, there was a decrease in active caspase-3 in the human meningioma cells, those that overexpressed MDK [28][39]. As an anti-apoptotic molecule, MDK prevents the natural defense system of the cell, further increasing cell growth and promoting MDK expression in tumor tissue [29][40]. MDK is also shown to impart cytoprotection to the Wilms’ tumor cells against cisplatin via the up-regulation of Bcl-2 proteins [30][41].
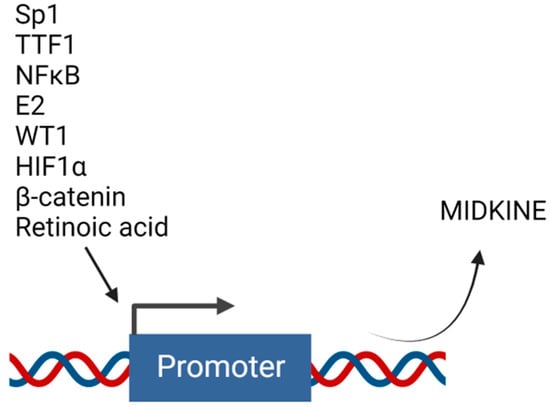
4. Transcriptional Regulators of MDK Expression
Several studies have described the role of MDK in drug resistance. However, not much has been completed to elucidate the mechanisms of regulation of MDK’s expression. Transcriptional regulation is necessary for the production of any protein. The transcriptional machinery is controlled by transcription factors that bind to the gene’s promoter region to transcribe the respective gene product. MDK is found to be regulated by various transcriptional regulators (Figure 4).Figure 4. Upstream transcription factors that regulate midkine gene expression. Transcription of MDK can be induced by different molecules that act as transcription factors and bind to the promoter region of MDK. Created with BioRender.com.
4.1. Estradiol
Estradiol (E2) is reported to elevate MDK mRNA expression in lung adenocarcinoma cells LTEP-a2 and A549 in a time-dependent manner. ICI 182,780 and tamoxifen, estrogen receptor (ER) inhibitors, inhibited the MDK expression induced by E2. In contrast, the antagonists for phosphoinositide-3 kinase and MAPK could not inhibit the same, suggesting the regulation of MDK by E2 at the transcriptional level. E2-induced Erβ was recruited to the estrogen response element in the MDK promoter. MDK regulates EMT, which plays a significant role in the migration of tumor cells in NSCLC. Knockdown of MDK showed a halt in EMT under E2 stimulation. MDK is an essential component of the E2-mediated EMT axis in NSCLC [31][25].
4.2. HIF-1α
Hypoxia inside the tumor is a barrier that tumor tissue needs to mitigate to flourish inside the system. Two potential Hypoxia Response Elements (HREs) exist in the MDK gene promoter region. HIF-1α induces MDK expression during hypoxic conditions, increasing the vascularization of the small pulmonary arteries and paving the vasculature [32][61], providing the basic regulatory elements needed for the cancer cells to survive.
4.3. NF-κB
The promoter region of MDK has a functional binding site for nuclear factor-κB (NF-κB) [33][62]. This explains the overexpression of MDK during inflammatory conditions. In prostate cancer cells, TNF-α was found to up-regulate the NF-κB pathway, thereby inducing the transcription of MDK [17][29].
4.4. SP1
The transcription factor, specificity protein 1 is encoded by the gene SP1. This protein is essential during the embryonic and early postnatal period [34][63]. Similar to the altered expression of MDK during inflammation and cancer, expression of SP1 is also elevated in human glioma tissues more than in normal tissue. SP1 binds to the putative transcription site in the promoter region of MDK and directly results in the upregulation of the same. This interaction is found to have cooperated in the tumorigenesis and progression of glioma [35][64].
4.5. TTF-1
The thyroid transcription factor (TTF) -1 regulates lung parenchyma formation and gene expression. TTF-1 binds to the TTF regulatory elements present in the 5′-region of the MDK promoter, and MDK expression was absent in the lungs of TTF-null mice. It is observed that TTF-1 is required for pulmonary MDK expression but not for its expression in non-pulmonary tissues. Other transcription factors may control the regulation of MDK in other tissues [36][65].
4.6. Retinoic Acid
Retinoic acid is a metabolite of Vitamin A and is needed by the body for normal growth and development. Retinoic acid regulates transcription by interacting with nuclear retinoic acid receptors (RARs) that are bound to retinoic acid response elements (RARE) near the target genes [37][66]. The human MDK gene has an upstream site that responds to Retinoic acid [38][67].
4.7. Beta Catenin
Beta-catenin is the key player of the Wnt-mediated pathway involved in the cells’ proliferation, differentiation, and apoptosis. Knockdown of MDK substantially downregulates the carcinogenic properties exhibited by Beta-catenin, a multifunctional protein reported to act as a transcription factor. Beta-catenin has been shown to bind to the promoter region of MDK and regulate it transcriptionally [39][68].
4.8. WT1
Wilms’ tumor is an embryonal malignancy of the kidney, and MDK is expressed in high levels during mid-gestation for developmental regulation of the embryo. The tumor suppressor gene for Wilms’ tumor is WT1. A study has shown that all Wilms’ tumor samples studied have a high level of MDK, and there are two WT1 elements in the human MDK promoter region. They have suggested that MDK is a target gene of WT1 [40][69].