The majority of pancreatic cancer patients have a poor prognosis, where the five-year survival rate is 9% in the United States, with an increasing incidence rate of 1.03% per year. Pancreatic ductal adenocarcinoma (PDAC) is an aggressive form of pancreatic cancer that makes up 90% of all diagnosed cases of pancreatic cancer. Other types of pancreatic cancer, such as neuroendocrine tumors, which secrete insulin, or acinar carcinomas, which release digestive enzymes, are less common. PDAC develops from neoplasms of the cells lining the pancreatic ducts and usually presents without symptoms until advanced stages of the disease. Here we discuss strategies for immunomodulation of pancreatic cancer.
- Pancreatic cancer
- microbiome
- T-cells
- Pancreatic cancer, microbiome, T-cells,
1. Introduction
The majority of pancreatic cancer patients have a poor prognosis, where the five-year survival rate is 9% in the United States, with an increasing incidence rate of 1.03% per year [1,2]. Pancreatic ductal adenocarcinoma (PDAC) is an aggressive form of pancreatic cancer that makes up 90% of all diagnosed cases of pancreatic cancer [3]. Other types of pancreatic cancer, such as neuroendocrine tumors, which secrete insulin, or acinar carcinomas, which release digestive enzymes, are less common. PDAC develops from neoplasms of the cells lining the pancreatic ducts and usually presents without symptoms until advanced stages of the disease [4].
The majority of pancreatic cancer patients have a poor prognosis, where the five-year survival rate is 9% in the United States, with an increasing incidence rate of 1.03% per year[1][2]. Pancreatic ductal adenocarcinoma (PDAC) is an aggressive form of pancreatic cancer that makes up 90% of all diagnosed cases of pancreatic cancer[3]. Other types of pancreatic cancer, such as neuroendocrine tumors, which secrete insulin, or acinar carcinomas, which release digestive enzymes, are less common. PDAC develops from neoplasms of the cells lining the pancreatic ducts and usually presents without symptoms until advanced stages of the disease[4].
At this time, there are limited treatment options for patients with advanced and incurable PDAC, all of which are chemotherapy based. The FOLFIRINOX regimen (a combination of oxaliplatin, irinotecan, leucovorin, and fluorouracil) is often used as the first line of treatment for patients who are likely to be able to tolerate the relatively high toxicity [5,6]. The median overall survival for this regimen is 11.1 months [6]. Patients with comorbidities are often treated with gemcitabine alone or in combination with albumin-bound (nab)-paclitaxel with a median overall survival of only approximately 6.8 and 8.5 months, respectively [5,6,7]. Surgical resection remains the only potentially curative option, with a median overall survival of 54.4 months when combined with adjuvant FOLFIRINOX [8]. However, only about 20% of patients are considered resectable at the time of diagnosis [9].
At this time, there are limited treatment options for patients with advanced and incurable PDAC, all of which are chemotherapy based. The FOLFIRINOX regimen (a combination of oxaliplatin, irinotecan, leucovorin, and fluorouracil) is often used as the first line of treatment for patients who are likely to be able to tolerate the relatively high toxicity[5][6]. The median overall survival for this regimen is 11.1 months[6]. Patients with comorbidities are often treated with gemcitabine alone or in combination with albumin-bound (nab)-paclitaxel with a median overall survival of only approximately 6.8 and 8.5 months, respectively[5][6][7]. Surgical resection remains the only potentially curative option, with a median overall survival of 54.4 months when combined with adjuvant FOLFIRINOX[8]. However, only about 20% of patients are considered resectable at the time of diagnosis[9].
Immunotherapy with immune checkpoint blockade (ICB) has been a breakthrough in treating some of the most treatment-refractory cancers, including advanced melanoma and metastatic non-small-cell lung carcinoma [10,11]. However, ICBs did not show efficacy in PDAC, and tumors showed primary resistance to monotherapy with ipilimumab (an anticytotoxic T lymphocyte antigen 4) or atezolizumab (anti-programmed cell death receptor ligand 1) [10,12]. Currently, the only approved immunotherapy for pancreatic cancer is pembrolizumab (anti-PD1), recommended for use in patients with mismatch repair deficiencies (dMMR) or microsatellite instability (MSI)-high tumors as a second line of treatment [13,14]. MSI-high tumors often have a higher tumor mutation burden and a more T-cell-inflamed microenvironment that is not typically seen in the immune-hostile PDAC tumor microenvironment. Modifying the tumor microenvironment to create a more T-cell-inflamed microenvironment might improve PDAC response to immunotherapy, either alone or in combination with other therapies.
Immunotherapy with immune checkpoint blockade (ICB) has been a breakthrough in treating some of the most treatment-refractory cancers, including advanced melanoma and metastatic non-small-cell lung carcinoma[10][11]. However, ICBs did not show efficacy in PDAC, and tumors showed primary resistance to monotherapy with ipilimumab (an anticytotoxic T lymphocyte antigen 4) or atezolizumab (anti-programmed cell death receptor ligand 1)[10][12]. Currently, the only approved immunotherapy for pancreatic cancer is pembrolizumab (anti-PD1), recommended for use in patients with mismatch repair deficiencies (dMMR) or microsatellite instability (MSI)-high tumors as a second line of treatment[13][14]. MSI-high tumors often have a higher tumor mutation burden and a more T-cell-inflamed microenvironment that is not typically seen in the immune-hostile PDAC tumor microenvironment. Modifying the tumor microenvironment to create a more T-cell-inflamed microenvironment might improve PDAC response to immunotherapy, either alone or in combination with other therapies.
2. Adjuvant and Neoadjuvant Therapies
Surgical resection is the best curative option for PDAC patients, although ultimately, 80% of those who undergo surgery recur, either due to micrometastases or microscopic residual disease in the tumor bed[1][15]. The current standard of care for upfront resectable PDAC is surgery followed by adjuvant combination chemotherapy[16]. The administration of gemcitabine and capecitabine after surgery resulted in median overall survival of 28.0 months, a modest increase compared to gemcitabine monotherapy with a median overall survival of 25.5 months[17]. The majority of patients in this trial had lymph node involvement. The efficacy of immunotherapy has also been tested in both neoadjuvant and adjuvant settings in other tumor types. Neoadjuvant immunotherapy has shown great promise primarily by reinvigorating exhausted T-cells before surgery. Tumor-specific CD8
Surgical resection is the best curative option for PDAC patients, although ultimately, 80% of those who undergo surgery recur, either due to micrometastases or microscopic residual disease in the tumor bed [1,50]. The current standard of care for upfront resectable PDAC is surgery followed by adjuvant combination chemotherapy [51]. The administration of gemcitabine and capecitabine after surgery resulted in median overall survival of 28.0 months, a modest increase compared to gemcitabine monotherapy with a median overall survival of 25.5 months [52]. The majority of patients in this trial had lymph node involvement. The efficacy of immunotherapy has also been tested in both neoadjuvant and adjuvant settings in other tumor types. Neoadjuvant immunotherapy has shown great promise primarily by reinvigorating exhausted T-cells before surgery. Tumor-specific CD8
+ T-cells can be primed by antigens derived from the primary tumor and can continue to exert their cytotoxic effects on micrometastases once the primary tumor is removed[18]. While univariate analysis showed that neoadjuvant immunotherapy resulted in better overall survival than adjuvant immunotherapy in pancreatic cancer patients, multivariate analysis showed that those treatment strategies were not significantly different[19].
T-cells can be primed by antigens derived from the primary tumor and can continue to exert their cytotoxic effects on micrometastases once the primary tumor is removed [53]. While univariate analysis showed that neoadjuvant immunotherapy resulted in better overall survival than adjuvant immunotherapy in pancreatic cancer patients, multivariate analysis showed that those treatment strategies were not significantly different [54].
Interestingly, adjuvant immunotherapy combined with chemotherapy was more effective than chemotherapy alone[20]. The combination may be effective, as chemotherapy can ablate tumors and release tumor antigens in the process. These antigens can then prime T-cells. Furthermore, treatment strategies reducing desmoplasia allows T-cells to infiltrate the tumor microenvironment more effectively[21]. Administration of nab-paclitaxel was found to reduce the density of the desmoplastic stroma in preclinical models and facilitated the delivery of gemcitabine to the tumor[22].
Interestingly, adjuvant immunotherapy combined with chemotherapy was more effective than chemotherapy alone [55]. The combination may be effective, as chemotherapy can ablate tumors and release tumor antigens in the process. These antigens can then prime T-cells. Furthermore, treatment strategies reducing desmoplasia allows T-cells to infiltrate the tumor microenvironment more effectively [23]. Administration of nab-paclitaxel was found to reduce the density of the desmoplastic stroma in preclinical models and facilitated the delivery of gemcitabine to the tumor [56].
The combination of radiation and chemotherapy was also effective in stimulating an anti-tumor immune response. Neoadjuvant chemoradiation given as 30 Gy of radiation combined with gemcitabine and a fluoropyrimidine derivative was found to increase the infiltration of both CD4
+
and CD8
+
T-cells. As expected, increased CD8
+ T-cell infiltration of the tumors was associated with prolonged overall survival and lower recurrence rates in patients with resectable pancreatic cancer[23]. Another study assessing the effect of neoadjuvant chemoradiation showed no difference in the recruitment of CD4
T-cell infiltration of the tumors was associated with prolonged overall survival and lower recurrence rates in patients with resectable pancreatic cancer [57]. Another study assessing the effect of neoadjuvant chemoradiation showed no difference in the recruitment of CD4
+
or CD8
+
T-cells; however, a decrease in FOXP3
+ Tregs was observed[24]. More studies are required to determine if T-cells’ recruitment can be further improved by optimizing the timing of treatment and surgery.
Tregs was observed [58]. More studies are required to determine if T-cells’ recruitment can be further improved by optimizing the timing of treatment and surgery.
3. Combination Strategies
PDAC is a complex disease with several cellular components that can either hinder or promote anti-tumor immunity. Therefore, successful therapeutic strategies may combine several existing therapies. For example, a combination of different ICBs to chemotherapy has shown success in lung cancer. Chemotherapy can induce apoptosis of tumor cells, and in the process, release tumor antigens that can activate anti-tumor immunity. A randomized phase II trial (NCT0287931) compared the use of gemcitabine and nab-paclitaxel against gemcitabine and nab-paclitaxel combined with durvalumab (anti-PDL1) and tremelimumab (anti-CTLA-4). Of the 11 patients that received gemcitabine, nab-paclitaxel, durvalumab, and tremelimumab, 8 achieved a partial response, showing the potential of combination therapy[25]. A CD40 agonist is being combined with gemcitabine, nab-paclitaxel, and nivolumab (anti-PD1) in a phase 1b clinical trial (NCT02482168) in patients with metastatic PDAC. Of 24 patients, 14 had a partial response[26]. Currently, this trial has progressed to a phase II study (NCT03214250).
PDAC is a complex disease with several cellular components that can either hinder or promote anti-tumor immunity. Therefore, successful therapeutic strategies may combine several existing therapies. For example, a combination of different ICBs to chemotherapy has shown success in lung cancer. Chemotherapy can induce apoptosis of tumor cells, and in the process, release tumor antigens that can activate anti-tumor immunity. A randomized phase II trial (NCT0287931) compared the use of gemcitabine and nab-paclitaxel against gemcitabine and nab-paclitaxel combined with durvalumab (anti-PDL1) and tremelimumab (anti-CTLA-4). Of the 11 patients that received gemcitabine, nab-paclitaxel, durvalumab, and tremelimumab, 8 achieved a partial response, showing the potential of combination therapy [59]. A CD40 agonist is being combined with gemcitabine, nab-paclitaxel, and nivolumab (anti-PD1) in a phase 1b clinical trial (NCT02482168) in patients with metastatic PDAC. Of 24 patients, 14 had a partial response [60]. Currently, this trial has progressed to a phase II study (NCT03214250).
4. T-Cell Activation Strategies for PDAC Treatment
With the modest progress made with chemotherapy, radiation, and ICBs, novel treatment strategies for pancreatic cancer are actively being explored. Considering that CD8
+
T-cells play a large role in the long-term survival of PDAC patients, approaches often involve activation of the anti-tumor immune response.
4.1. Chimeric Antigen Receptor (CAR) T-Cells
CAR T-cell therapy is an adoptive cell therapy, where autologous T-cells are isolated and genetically engineered to express receptors that bind to tumor antigens. These modified T-cells are then reinfused into the patient. When CAR T-cells recognize target antigens, they become activated and exert cytotoxic effects on the tumor cell (Figure 1a). Currently, CAR T-cell therapy has only been approved by the FDA for hematologic malignancies[27]. The safety of a mesothelin-specific CAR T-cell therapy was tested in six patients with chemotherapy-refractory metastatic PDAC. These patients did not experience a cytokine storm or dose-limiting toxicities. However, only two patients achieved stable disease with overall survival of 3.8 and 5.4 months[28]. Preclinical studies are also testing the efficacy of switchable CAR T-cells against human epithelial growth factor receptor 2 (HER2) antigen. Switchable CAR T-cells are composed of two parts. For the first part, the autologous T-cells are engineered with the signaling portion of the T-cell receptor and the second part consists of a “switch” protein that binds to both the signaling portion of the autologous T-cells and the tumor antigen. This allows the fine-tuning of the T-cell response by administering variable concentrations of the switch protein. The switch CAR T-cell against HER2 antigen led to the complete remission of xenograft models derived from patients with stage IV PDAC[29].
CAR T-cell therapy is an adoptive cell therapy, where autologous T-cells are isolated and genetically engineered to express receptors that bind to tumor antigens. These modified T-cells are then reinfused into the patient. When CAR T-cells recognize target antigens, they become activated and exert cytotoxic effects on the tumor cell (Figure 1a). Currently, CAR T-cell therapy has only been approved by the FDA for hematologic malignancies [61]. The safety of a mesothelin-specific CAR T-cell therapy was tested in six patients with chemotherapy-refractory metastatic PDAC. These patients did not experience a cytokine storm or dose-limiting toxicities. However, only two patients achieved stable disease with overall survival of 3.8 and 5.4 months [62]. Preclinical studies are also testing the efficacy of switchable CAR T-cells against human epithelial growth factor receptor 2 (HER2) antigen. Switchable CAR T-cells are composed of two parts. For the first part, the autologous T-cells are engineered with the signaling portion of the T-cell receptor and the second part consists of a “switch” protein that binds to both the signaling portion of the autologous T-cells and the tumor antigen. This allows the fine-tuning of the T-cell response by administering variable concentrations of the switch protein. The switch CAR T-cell against HER2 antigen led to the complete remission of xenograft models derived from patients with stage IV PDAC [63].
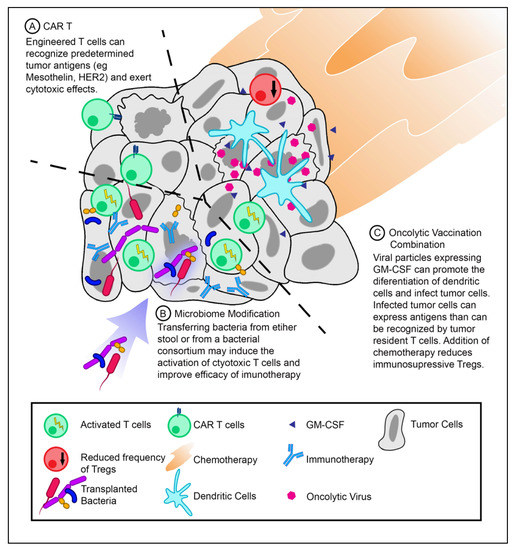
Figure 1.
T-cell activation strategies for pancreatic ductal adenocarcinoma (PDAC) treatment. (
A
) Infusion of chimeric antigen receptor T-cells (CAR T-cells) against PDAC tumor antigens. CAR T-cells can recognize antigens specific for their CARs and exert cytotoxic effects. (
B
) Modifying the tumor microbiome may introduce novel antigens that can activate T-cells and improve the efficacy of immunotherapeutics. (
C
) Therapeutic vaccination with oncolytic viruses expressing granulocyte–macrophage colony-stimulating factor (GM-CSF) in combination with chemotherapy (low-dose cyclophosphamide) can increase the frequency of dendritic cells while decreasing the frequency of Tregs.
Unfortunately, engineered T-cells become progressively dysfunctional within the tumor and require repeated reinfusions to achieve a therapeutic benefit in experimental models[30]. One study attempted to mitigate this process by modulating TAM function. The administration of agonistic anti-CD40 along with CAR T-cells resulted in increased proliferation and cytotoxic abilities of CAR T-cells[31]. Notably, while the depletion of TAMs resulted in increased proliferation of endogenous T-cells, there was no marked increase in CAR T-cell infiltration of the tumor[31]. The efficacy of CAR T-cell function can also be improved by coexpressing an engineered immunomodulatory fusion protein. A fusion protein consisting of the inhibitory Fas receptor ectodomain and a stimulatory 4-1BB endodomain could convert inhibitory signals into activating ones. The use of CAR T-cells expressing the Fas-4-1BB fusion protein was found to significantly improve survival in an aggressive mouse model of pancreatic cancer[32]. This suggests that several methods can be developed to improve the efficacy of current CAR T-cell technology to treat pancreatic cancers.
Unfortunately, engineered T-cells become progressively dysfunctional within the tumor and require repeated reinfusions to achieve a therapeutic benefit in experimental models [64]. One study attempted to mitigate this process by modulating TAM function. The administration of agonistic anti-CD40 along with CAR T-cells resulted in increased proliferation and cytotoxic abilities of CAR T-cells [65]. Notably, while the depletion of TAMs resulted in increased proliferation of endogenous T-cells, there was no marked increase in CAR T-cell infiltration of the tumor [65]. The efficacy of CAR T-cell function can also be improved by coexpressing an engineered immunomodulatory fusion protein. A fusion protein consisting of the inhibitory Fas receptor ectodomain and a stimulatory 4-1BB endodomain could convert inhibitory signals into activating ones. The use of CAR T-cells expressing the Fas-4-1BB fusion protein was found to significantly improve survival in an aggressive mouse model of pancreatic cancer [66]. This suggests that several methods can be developed to improve the efficacy of current CAR T-cell technology to treat pancreatic cancers.
4.2. Microbiome Modification
As mentioned previously, the tumor microbiome can predict the overall survival of PDAC patients. This suggests that modification of the microbiome can potentially increase the response to therapeutics. The microbiome can be altered either by antibiotics, FMT, or defined bacterial consortia (Figure 1b).
4.2.1. Antibiotics
Recently, the use of antibiotics has been shown to abrogate the effects of anti-PD1 immunotherapy in both melanoma and renal cell carcinoma[33]. However, the opposite was found to be true by Pushalkar et al.[34] in regard to PDAC. An increase in bacteria was found in the cancerous pancreas of mice compared to healthy pancreas. Decreasing bacterial abundance using broad-spectrum antibiotics was successful in slowing tumor growth in KC mice (spontaneous pancreatic neoplasia model). This growth was further attenuated by combining broad-spectrum antibiotics with anti-PD1. The use of antibiotics was found to increase the intratumoral CD8/CD4 T-cell ratio, which may explain the reduction in tumor growth (Figure 2a)[34]. Antibiotic treatment strategies could be employed in neoadjuvant settings to abrogate harmful bacteria before surgery. More studies are required to validate this strategy.
Recently, the use of antibiotics has been shown to abrogate the effects of anti-PD1 immunotherapy in both melanoma and renal cell carcinoma [45]. However, the opposite was found to be true by Pushalkar et al. [47] in regard to PDAC. An increase in bacteria was found in the cancerous pancreas of mice compared to healthy pancreas. Decreasing bacterial abundance using broad-spectrum antibiotics was successful in slowing tumor growth in KC mice (spontaneous pancreatic neoplasia model). This growth was further attenuated by combining broad-spectrum antibiotics with anti-PD1. The use of antibiotics was found to increase the intratumoral CD8/CD4 T-cell ratio, which may explain the reduction in tumor growth (Figure 2a) [47]. Antibiotic treatment strategies could be employed in neoadjuvant settings to abrogate harmful bacteria before surgery. More studies are required to validate this strategy.
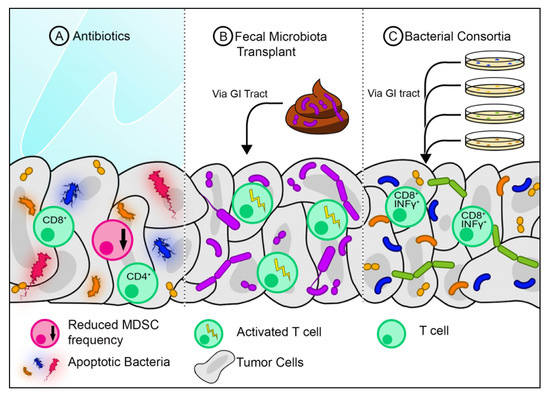
Figure 2.
A
+
+
B
C
+
4.2.2. Fecal Microbiota Transplantation
FMT is commonly used to treat dysbiosis of the gut with a high success rate, such as with refractory Clostridium difficile infections. Presumably, FMT can be used to treat other conditions with microbial dysbiosis, including PDAC[35][36]. Akkermansia muciniphila, a commensal bacterium commonly found in healthy stool, was found to induce increased infiltration of CD4
FMT is commonly used to treat dysbiosis of the gut with a high success rate, such as with refractory Clostridium difficile infections. Presumably, FMT can be used to treat other conditions with microbial dysbiosis, including PDAC [44,67]. Akkermansia muciniphila, a commensal bacterium commonly found in healthy stool, was found to induce increased infiltration of CD4
+ T-cells in RET melanomas when combined with the anti-PD1 antibody[33]. This suggests that certain bacteria found in healthy stool samples may boost immunity by activating certain populations of T-cells. Modifying the gut microbiome via FMT may also be an attractive strategy in the neoadjuvant setting for PDAC (Figure 2b).
T-cells in RET melanomas when combined with the anti-PD1 antibody [45]. This suggests that certain bacteria found in healthy stool samples may boost immunity by activating certain populations of T-cells. Modifying the gut microbiome via FMT may also be an attractive strategy in the neoadjuvant setting for PDAC (Figure 2b).
4.2.3. Bacterial Consortia
The gut is colonized by various bacterial populations, some of which have a capacity to induce T-cell activation (Figure 2c). A consortium of 11 bacteria was found to increase the frequency of INFγ
+
-producing CD8
+ T-cells in the intestines of germ-free mice. This consortium consisted of seven Bacteroidales and four non-Bacteroidales species, all of which needed to be administered in combination to achieve maximal T-cell activation. When administered with an anti-PD1 or anti-CTLA 4, the mix of 11 bacteria slowed tumor growth in mice[37]. This suggests that a mix of bacteria may be taken as a probiotic to improve the efficacy of immunotherapy. Whether this would benefit PDAC patients still needs to be explored.
T-cells in the intestines of germ-free mice. This consortium consisted of seven Bacteroidales and four non-Bacteroidales species, all of which needed to be administered in combination to achieve maximal T-cell activation. When administered with an anti-PD1 or anti-CTLA 4, the mix of 11 bacteria slowed tumor growth in mice [68]. This suggests that a mix of bacteria may be taken as a probiotic to improve the efficacy of immunotherapy. Whether this would benefit PDAC patients still needs to be explored.
4.3. Oncolytic Vaccine Combinations
4.3. Oncolytic Vaccine Combinations
Oncologic vaccines work to expose the host immune system to tumor-specific antigens. To date, vaccines on their own have shown no clinical benefits in pancreatic cancer [69]. However, there are some promising results indicative of inducing an anti-tumor immune response when vaccines are combined with chemotherapy. An irradiated, granulocyte-macrophage colony-stimulating factor (GM-CSF), secreting, allogeneic PDAC vaccine (GVAX) was given as a single agent or in combination with low-dose cyclophosphamide to patients with resectable PDAC. After two weeks of vaccine administration, tertiary lymphoid organs were found inside tumor biopsies. This indicates that lymphocytes are actively infiltrating the tumor microenvironment and facilitating an adaptive response [70].
Reference
Oncologic vaccines work to expose the host immune system to tumor-specific antigens. To date, vaccines on their own have shown no clinical benefits in pancreatic cancer[38]. However, there are some promising results indicative of inducing an anti-tumor immune response when vaccines are combined with chemotherapy. An irradiated, granulocyte-macrophage colony-stimulating factor (GM-CSF), secreting, allogeneic PDAC vaccine (GVAX) was given as a single agent or in combination with low-dose cyclophosphamide to patients with resectable PDAC. After two weeks of vaccine administration, tertiary lymphoid organs were found inside tumor biopsies. This indicates that lymphocytes are actively infiltrating the tumor microenvironment and facilitating an adaptive response[39].
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA. Cancer J. Clin. 2019, 69, 7–34.
- Saad, A.M.; Turk, T.; Al-Husseini, M.J.; Abdel-Rahman, O. Trends in pancreatic adenocarcinoma incidence and mortality in the United States in the last four decades; A SEER-based study. BMC Cancer 2018, 18, 1–11.
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 1–20.
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 1–23.
- Muñoz Martín, A.J.; Adeva, J.; Martínez-Galán, J.; Reina, J.J.; Hidalgo, M. Pancreatic ductal adenocarcinoma: Metastatic disease. Clin. Transl. Oncol. 2017, 19, 1423–1429.
- Vaccaro, V.; Sperduti, I.; Milella, M. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 365, 768–769.
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703.
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 2018, 379, 2395–2406.
- He, J.; Ahuja, N.; Makary, M.A.; Cameron, J.L.; Eckhauser, F.E.; Choti, M.A.; Hruban, R.H.; Pawlik, T.M.; Wolfgang, C.L. 2564 resected periampullary adenocarcinomas at a single institution: Trends over three decades. Hpb 2014, 16, 83–90.
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465.
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454.
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent ipilimumab (Anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833.
- Sohal, D.P.S.; Kennedy, E.B.; Khorana, A.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Krishnamurthi, S.; Moravek, C.; O’Reilly, E.M.; Philip, P.A.; et al. Metastatic pancreatic cancer: ASCO clinical practice guideline update. J. Clin. Oncol. 2018, 36, 2545–2556.
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; de Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/ mismatch repair–deficient cancer: Results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 2020, 38, 1–10.
- Li, D.; Xie, K.; Wolff, R.; Abbruzzese, J.L. Pancreatic cancer. Lancet 2004, 363, 1049–1057.
- Seufferlein, T.; Ettrich, T.J. Treatment of pancreatic cancer—neoadjuvant treatment in resectable pancreatic cancer (PDAC). Transl. Gastroenterol. Hepatol. 2019, 4.
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024.
- O’Donnell, J.S.; Hoefsmit, E.P.; Smyth, M.J.; Blank, C.U.; Teng, M.W.L. The promise of neoadjuvant immunotherapy and surgery for cancer treatment. Clin. Cancer Res. 2019, 25, 5743–5751.
- Amin, S.; Baine, M.; Meza, J.; Lin, C. The impact of neoadjuvant and adjuvant immunotherapy on the survival of pancreatic cancer patients: A retrospective analysis. BMC Cancer 2020, 20, 1–10.
- Tran, T.B.; Maker, V.K.; Maker, A.V. Impact of Immunotherapy after Resection of Pancreatic Cancer. J. Am. Coll. Surg. 2019, 229, 19–27.e1.
- Von Hoff, D.D.; Ramanathan, R.K.; Borad, M.J.; Laheru, D.A.; Smith, L.S.; Wood, T.E.; Korn, R.L.; Desai, N.; Trieu, V.; Iglesias, J.L.; et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: A phase I/II trial. J. Clin. Oncol. 2011, 29, 4548–4554.
- Homma, Y.; Taniguchi, K.; Murakami, T.; Nakagawa, K.; Nakazawa, M.; Matsuyama, R.; Mori, R.; Takeda, K.; Ueda, M.; Ichikawa, Y.; et al. Immunological impact of neoadjuvant chemoradiotherapy in patients with borderline resectable pancreatic ductal adenocarcinoma. Ann. Surg. Oncol. 2014, 21, 670–676.
- Tsuchikawa, T.; Hirano, S.; Tanaka, E.; Matsumoto, J.; Kato, K.; Nakamura, T.; Ebihara, Y.; Shichinohe, T. Novel aspects of preoperative chemoradiation therapy improving anti-tumor immunity in pancreatic cancer. Cancer Sci. 2013, 104, 531–535.
- Renouf, D.J.; Dhani, N.C.; Kavan, P.; Jonker, D.J.; Wei, A.C.; Tang, P.A.; Graham, B.; Gallinaro, L.; Hasan, T.; Li, W.; et al. The Canadian Cancer Trials Group PA.7 trial: Results from the safety run in of a randomized phase II study of gemcitabine (GEM) and nab-paclitaxel (Nab-P) versus GEM, nab-P, durvalumab (D), and tremelimumab (T) as first-line therapy in metastatic pancreat. J. Clin. Oncol. 2018, 36, 349.
- O’Hara, M.H.; O’Reilly, E.M.; Rosemarie, M.; Varadhachary, G.; Wainberg, Z.A.; Ko, A.; Fisher, G.A.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. Abstract CT004: A Phase Ib study of CD40 agonistic monoclonal antibody APX005M together with gemcitabine (Gem) and nab-paclitaxel (NP) with or without nivolumab (Nivo) in untreated metastatic ductal pancreatic adenocarcinoma (PDAC) patients. AACR 2019, 79, CT004.
- Date, V.; Nair, S. Emerging vistas in CAR T-cell therapy: Challenges and Opportunities in Solid Tumors. Expert Opin. Biol. Ther. 2020, 1–16.
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32.
- Raj, D.; Yang, M.H.; Rodgers, D.; Hampton, E.N.; Begum, J.; Mustafa, A.; Lorizio, D.; Garces, I.; Propper, D.; Kench, J.G.; et al. Switchable CAR-T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut 2019, 68, 1052–1064.
- Stromnes, I.M.; Schmitt, T.M.; Hulbert, A.; Brockenbrough, J.S.; Nguyen, H.; Cuevas, C.; Dotson, A.M.; Tan, X.; Hotes, J.L.; Greenberg, P.D.; et al. T cells Engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Physiol. Behav. 2016, 176, 139–148.
- Stromnes, I.M.; Burrack, A.L.; Hulbert, A.; Bonson, P.; Brockenbrough, J.S.; Raynor, J.F.; Spartz, E.J.; Robert, H.; Greenberg, P.D.; Hingorani, S.R.; et al. Differential effects of depleting versus programming tumor-associated macrophages on engineered T cells in pancreatic ductal adenocarcinoma. Cancer Immunol. Res. 2020, 7, 977–989.
- Oda, S.K.; Anderson, K.G.; Ravikumar, P.; Bonson, P.; Garcia, N.M.; Jenkins, C.M.; Zhuang, S.; Daman, A.W.; Chiu, E.Y.; Bates, B.M.; et al. A Fas-4-1BB fusion protein converts a death to a pro-survival signal and enhances T cell therapy. J. Exp. Med. 2020, 217, e20191166.
- Chanyi, R.M.; Craven, L.; Harvey, B.; Reid, G.; Silverman, M.J.; Burton, J.P. Faecal microbiota transplantation: Where did it start? What have studies taught us? Where is it going? SAGE Open Med. 2017, 5, 205031211770871.
- Tanoue, T.; Morita, S.; Plichta, D.R.; Skelly, A.N.; Suda, W.; Sugiura, Y.; Narushima, S.; Vlamakis, H.; Motoo, I.; Sugita, K.; et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 2019, 565, 600–605.
- Beatty, G.L.; Eghbali, S.; Kim, R. Deploying Immunotherapy in Pancreatic Cancer: Defining Mechanisms of Response and Resistance. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 267–278.
- Lutz, E.R.; Wu, A.A.; Bigelow, E.; Sharma, R.; Mo, G.; Soares, K.; Solt, S.; Dorman, A.; Wamwea, A.; Yager, A.; et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol. Res. 2014, 2, 616–631.